

 PDF
PDF ePub
ePub Citation
Citation Print
Print


X-linked retinoschisis is one of the most common causes of juvenile macular degeneration in young males.1 A gene, called XLRS1, has been cloned and identified as cause of X-linked retinoschisis.2 The XLRS1 gene has six exons and encodes a 224 amino acid protein, processed by N-terminal cleavage into a mature protein with a calculated size of 23 kDa (201 amino acids). The predicted protein sequence contains a highly conserved motif implicated in cell-cell interaction and, thus, may be active in cell adhesion processes. Most mutations, predominantly missense, were found in exons 4-6 encoding this discoidin domain.3
Here we present observational case report of a novel missense mutation in the XLRS1 gene in a family of Korean patients that confirmed the diagnosis of X-linked retinoschisis.
Case Report
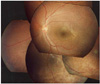
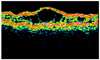
A 9 year old boy was found to have poor visual acuity. His best corrected visual acuity was 0.3 in both eyes. Refractions were OD: +1.25, OS: +1.00. Fundus examination showed bilateral wheel-like maculopathy with a golden-yellow reflex at the inferior peripheral fundus (Fig. 1). Single-flash ERG documented marked reduction of b wave (Fig. 2). Visual fields showed paracentral scotomas in both eyes. Color vision test was normal with the Ishihara's test. Optical coherence Tomography confirmed foveal schisis (Fig. 3).
During examination, his maternal uncle was also diagnosed as X-linked retinoschisis. His best corrected visual acuity was 0.025 in the right eye and 0.4 in the left. His fundus also showed typical cystic maculopathy.
After informed consent, we performed complete ophthalmologic and molecular genetic study of all available family members of the affected boy.
Genomic DNA was isolated from peripheral blood samples. All exons of the XLRS1 gene were amplified by polymerase chain reaction and analyzed using a direct sequencing method. Primers were designed with using Genetix software (Software development). Searches for information and homology of nucleotide and amino acid sequence was analyzed using homology search with BLAST against the sequences in the Gene Bank and EMBL DNA database.
Direct sequencing showed that two cytosine residues were substituted into thymines and coded Phenylalanine instead of Leucine (Leu103Phe) in exon 4 of XLRS1 gene (Fig. 4). This mutations were also identified in his sister, mother and maternal uncle. The hemizygous mutation was confirmed using both sense and antisense primers.
Discussion
We investigated both ophthalmological and molecular genetic study of families of 3 unrelated probands. All mutations were missense mutations in the exon 4 of XLRS1 gene. Two were known already (Arg102Trp, and Trp96Arg). And we found a novel missense mutation of Leu103Phe. We are unware of previous reports of this mutation and could find no reference to it in a computerized search utilizing MEDLINE and website (http://www.dmd.nl/rs/index). As with other mutations, this mutation also occurred in the discoidin domain.
The clinical feature of the boy and maternal uncle is mainly cystic maculopathy. The analysis of the function of the different forms of mutation in XLRS1 gene may be needed to enhance our understanding of X-linked retinoschisis pathogenesis.


 XML Download
XML Download