

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Alport syndrome is a basement membrane disorder characterized clinically by hereditary nephropathy, sensorineural hearing loss and ocular abnormalities.1-5 It has been associated with X-linked, autosomal recessive and autosomal dominant inheritance.6,7 The X-linked mutations have been mapped to defects in the α-5-chain of the type IV collagen (COL4A5) gene, subsequently also compromising COL4A3 and COL4A4.8-11 All reported mutations lead to abnormalities in the basement membranes of the glomerulus, cochlea, retina, lens capsule and cornea, which eventually contribute to the typical phenotypes of the syndrome.4,5
Ocular features most frequently described include lens abnormalities, characterized by lenticonus,4 cataract,5 retinal flecks,12,13 macular hole,14 and corneal arcus.6 Brownell and Wolter15 have demonstrated the thinning of the lens capsule by light microscopy. Electron microscopic study has demonstrated the marked thinning and vertical dehiscence of the anterior lens capsule in Alport syndrome.16,17
Here, we report the detailed ultrastructural findings in the four lens capsules obtained from two Alport syndrome patients at the time of surgery.
CASE REPORT
1. Clinical history
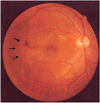
A 31-year-old man visited our clinic on January 31, 2002, complaining of decreased vision in both eyes. Gross examination did not show any abnormal findings except short stature. His best-corrected visual acuity (BCVA) was 20/50 in each eye with a -13.00 sphere OD and a -12.00 sphere OS. Retinoscopy showed oil-globule reflex in both eyes. Slit-lamp examination disclosed a central, nipple-like protrusion of the anterior lenticonus accompanied by a subcapsular faint opacity in both eyes (Fig. 1). Ophthalmoscopy revealed yellow punctate lesions in the retinal pigment epithelium (RPE) level of both eyes that spared the macula (Fig. 2). Fluorescein angiography demonstrated bilateral multiple window defects. Electroretinogram (ERG) and specular microscopy revealed normal findings.
He had first noted dark urine at the age of 18 years, at which time massive hematuria and proteinuria were found. At 21, he had developed irreversible renal failure, which eventually required hemodialysis. He had sensorineural hearing loss by audiometry.
An uncomplicated phacoemulsification with a continuous circular capsulorrhexis and foldable intraocular lens (IOL) implantation was performed in both eyes. The capsules were fragile during the capsulectomy. Postoperatively, BCVA was 20/20, and no postoperative complication was observed in either eye. We studied the ultrastructural findings in the anterior lens capsules obtained at the time of surgery.
2. Family history
The patient had three younger siblings. Both brothers (26 and 24 years) had Alport syndrome, had received hemodialysis, had anterior lenticonus, and were also deaf as determined by audiometry. The younger sister (21 years) was asymptomatic. The 26-year-old affected brother of two affected brothers, received clear lens extraction and foldable IOL implantation in our hospital. His anterior lens capsules also underwent ultrastructural examination. But, the 24-year-old affected brother of two affected brothers, received clear lens extraction and foldable IOL implantation in other hospital. So we could not accepted anterior lens capsules. No member of the third generation was symptomatic. The patien's 64-year-old mother, had hypertension but no known renal problem.
3. Ultrastructure of the anterior lens capsule
All four anterior lens capsules were fixed in 2.5% glutaraldehyde and processed in epoxy resin for transmission electron microscopy.
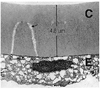
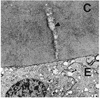
Thinning was found not only in the central part of the anterior capsule, but also in the peripheral part. The anterior lens capsular thickness varied from 4 to 13 µm, with the thickest portion on the periphery and the thinnest in the center (Fig. 3). The central thin portion contrasted sharply with the 18 µm control central capsule, the normal thickness at this age. The outer one-third of the capsule was looser and of fibrillar texture (Figs. 3, 5). The inner two-thirds of the capsule was remarkable for having innumerable dehiscences, beginning at this the inner surface and extending straight out or curving around to join other dehiscences (Figs. 3, 4). Almost every dehiscence ran vertically, but some ran horizontally. The vertical dehiscences varied from 1.0 to 7.0 µm in depth, and from 200 to 500 nm in width. The dehiscences were particularly prominent in the central part of the anterior capsule but they were also observed in the peripheral region. All dehiscences contained fibrillar material and vacuoles (Fig. 5).
Electron microscopy of the anterior lens epithelium showed some pathologic changes. The lens epithelium was highly irregular. The lens epithelial cells were highly irregular in shape and their lateral borders were indistinct. Their nuclei were smaller, darker, and irregular margined. The cytoplasm contained numerous lacunae. There was a paucity of other organelles (Fig. 6A). The lens epithelial cells were occasionally multilayered or dropout (Fig. 6B). The lens epithelial cells were well preserved in the focal area. The epithelial cells were cuboidal-shaped and their lateral borders were closly joined (Fig. 6C).
DISCUSSION
Histologic and ultrastructural examination of the kidney in Alport syndrome reveals characteristic, diffuse lamellation of the glomerular basement membrane.18 In the cochlea, there is atrophy of the capillary basement membranes of the stria vascularis.18 A possible defect of Bruch's membrane has been suggested for the pathogenesis of the retinal flecks.19
The capsular thinning and anterior bulging of the lens, as well as reports of spontaneous rupture, were suggestive of defective capsular strength.20,21 In the present study, the capsular thinning offered convincing evidence that the lens capsule in the eye had ruptured and fragmented during capsulectomy.
Although the pathogenesis of Alport syndrome is not precisely understood, it has recently become apparent that abnormalities in the type IV collagen molecules, which are a major structural component of the basement membrane, lead to the development of this syndrome.22,23 The basement membrane is composed of type IV collagen, laminin, and various heparan sulfate proteoglycans.24 Type IV collagen is thought to be the major structural element. This molecule was initially believed to be a heterotrimer containing two α-1(IV) chains and one α-2 chain. Recently, evidence for the existence of four additional different chains, α-3, α-4, α-5, and α-6, has been reported.25 Each chain contains three distinct domains: a carboxyl nontriple-helical (NC-1) domain, a triple helical (COL1) domain, and an amino terminal (7S) domain.25 In the basement membrane, type IV collagen forms a mesh structure with the NC-1 domain acting as hinges.26
In 1982, McCoy and colleagues,27 proposed the Good-pasture antigen to be the α-3(IV) NC-1 domain that is absent in the patient with Alport syndrome. Additionally, a point mutation in the type IV collagen α-5 chain gene has been identified in Alport syndrome. Accordingly, the X-linked form is thought to be caused by mutations in the type IV collagen α-5 chain gene (COL4A5),28 and subsequently COL4A3 and COL4A4 are also compromised.9-11 In patients with Alport syndrome, because of an abnormality in NC-1 domains, as in a frame structure without hinges, it becomes difficult to maintain the cubic structure.26 The basement membrane becomes thin and fragile and undergoes a mechanical stress associated accommodation. Gradually, dehiscences are produced and anterior lenticonus results.25
Streeten and colleagues29 reported ultrastructural findings in the anterior lens capsule from a 30-year-old Alport patient. Their observations included marked thinning of the anterior lens capsule and many dehiscences. These results agreed well with our own ultrastructural observations; namely, (1) almost every dehiscence ran vertically, (2) the dehiscences were located in the inner two-thirds of the anterior capsule, and (3) the dehiscences contained fibrillar material and vacuoles.
The marked involvement in the anterior polar region may be a response to the stresses of accommodation in the area where movement displacement is greatest and the lens is least supported.21 Lesser displacement occurs in the posterior lenticonus and cataract in this disease.5,24 The frequency of cellular debris under the capsule at the dehiscences may indicate that their formation is traumatic to the underlying cells, resulting in their rupture.16 This correlates with the irregular shape of epithelial cells seen in our patient, an observation also reported by others using light microscopy and one that could be directly related to the formation of anterior polar and superficial cortical cataract. On the basis of our findings, the lens epithelial abnormality coexisted with a lens capsular disorder. This could indicate that the lens epithelium might be also involved in this disease.
In the present case, it should be emphasized that the dehiscences never extended into the lens capsule and were limited to the inner two-thirds of the capsule. In composition and structure, the lens capsule is usually considered to consist of uniform tissues. However, based on the above findings, it is presumed that there are differences in the composition and structure between the inner and outer layers of the lens capsule. Our histologic examination in this study suggests that the primary abnormalities in Alport syndrome are limited to the inner layer of the lens capsule.
In patients with Alport syndrome, because of decreased visual acuity or the complications of cataract, it could be that lensectomy and IOL implantation may be indicated, making it very likely that the lens capsule will become even more fragile. During surgery, attention must be paid to the procedures of capsulotomy and lensectomy. In the above patient, the IOL was fixed within the capsule, and no complications were observed during or after operation.
Alport syndrome is most commonly transmitted as an X-linked dominant disease, although autosomal dominant and recessive patterns have been described. In our study, the presence of the syndrome in the patient's kindred was usual in X-linked dominant Alport disease. The male patients are more severely affected and usually die before 30 years of age. The difficulty in identifying affected female patients is illustrated by the asymptomatic female patient in the present family.
Further study will be needed to determine whether the molecular defect in this family involves exactly the some antigen as that in the recently reported cases.



 XML Download
XML Download