

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Sandhoff disease is a rare autosomal recessive disease caused by the deficiency of both lysosomal hydrolase β-hexosaminidase A and β-hexosaminidase B. The deficiency of these lysosomal isoenzymes leads to the accumulation of GM2 ganglioside and related glycolipids, particularly in neurons and viscera.1 This disorder exhibits considerable clinical heterogeneity and has three clinical forms (infantile, juvenile, adult) which differ according to the age of onset and severity.2
The infantile form of Sandhoff disease is characterized by the early onset of symptoms, which usually occur in the first 6 to 18 months of life. Clinical symptoms are severe, characterized by progressive neurologic impairment, generalized hypotonia, hyperacusis, tonic-clonic seizures, and bilateral cherry red spots in the macular region of the retina. This form usually presents a stereotypical progression of disease leading to death before the age of 4 years.3
The juvenile form of Sandhoff disease becomes symptomatic between the ages of 4 and 6 years, characterized by dementia. Typically, there are fewer severe manifestations, such as cerebellar ataxia, mental retardation, spinal muscular atrophy and local panatrophy, than in the infantile form of Sandhoff disease. Furthermore, these patients tend to die later, between childhood and early adulthood.4
The adult form of Sandhoff disease usually manifests neurologic signs and symptoms in early childhood, characterized by a protractive course. At least 35% of these patients have their first symptom before the age of 10. Progression of clinical signs and symptoms is usually slow. Although clinical phenotype of this disorder varies widely, spinocerebellar degeneration or motor neuron disease has often been reported.5-7 The cherry red spot in the macula is less frequently detected in this form.8
Recently, the report that the biochemical defect in Sandhoff disease is the result of a mutation in the gene on chromosome 5q13 coding the β subunit of β-hexosaminidase has been presented.9
To date, there have been no case reports in the literature of Sandhoff disease in Korea. We present here the first case report of a Korean patient with infantile form of Sandhoff disease, presenting typical neurologic impairment, bilateral optic atrophy, and a cherry red spot in the macula.
CASE REPORT
A 4 year old Korean girl who was being treated with anticonvulsants, had presented progressive physical and mental regression noted from the age of 1 year. She had been treated with anticonvulsants to alleviate her chronic tonic-clonic seizures, but symptoms were prolonged and uncontrollable. Since she could not follow objects with her eyes and did not have visual attention, she was referred to our ophthalmology clinic. Furthermore, she had been concurrently referred to a genetic and metabolic clinic because her neurologic symptoms progressed and she had a family history in which her older sister had died from the same symptoms.
The patient was born by vaginal delivery at full term gestational age and had a normal physical build. Both of her parents were clinically healthy and had a non specific disorder. However, her older sister had died from similar symptoms at 7 years old which had not been assessed accurately.
At the first ophtalmologic examination, she showed generalized hypotonia, quadriparesis, and a brisk deep tendon reflex. Ophthalmologic findings showed that she could not fixate her eyes on objects and could not follow moving targets. Moreover, the oculocephalic reflex and optokinetic nystagmus did not exist. Anterior segments of both eyes showed normal findings but there was a weak and sluggish pupillary response to light in both eyes. A pale optic disc and a cherry red spot in the macula were seen in both eyes by ophthalmoscope (Fig. 1A, 1B, 1C).
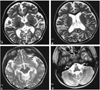
Low signal intensity at the bilateral thalamus and high signal intensity at the cerebral white matter were noted in a T2-weighted brain MR image (Fig. 2A, 2B). Cerebral cortex and cerebellum showed general atrophic changes (Fig. 2C, 2D).
Genetic and metabolic examinations, which were assessed by the Korean Genetics Research Center and the Mayo Medical Laboratories in the USA showed significant results. Lysosomal enzyme assay of peripheral blood revealed that there was both β-hexosaminidase A and B isoenzyme were absent in the serum. Further enzyme assay using fibroblasts showed a marked reduction of both total β-hexosaminidase A and B, at 1.8 U/g (reference range: 92.5 U/g to 184.5 U/g). In particular, the percentage of β-hexosaminidase A was elevated as 76% (reference range: 42% to 62%) (Table 1). The β-hexosaminidase system consists of two major isoenzymes A and B, and one minor isoenzyme S.10 Consequently, the percentage of β-hexosaminidase B was lower than 24% (reference range: 41% to 55%) (Table 1).
During the most recent follow up examination the patient still exhibited similar ophthalmic symptoms that we had noted previously, though she showed good seizure control, under anticonvulsant therapy. she did not show any progression of visual ability.
DISCUSSION
Sandhoff disease is caused by the deficiency of both lysosomal hydrolase β-hexosaminidase A and β-hexosaminidase B. Although the disease has an autosomal recessive transmissionis, a few sporadic casesforms have been reported.1
The β-hexosaminidase system consists of two major isoenzymes, β-hexosaminidase A (α-β) and β-hexosaminidase B (β-β) as well as one minor isoenzyme β-hexosaminidase S (α-α). These isoenzymes are formed by the different combinations of the two subunits, α and β.10 The α subunit is encoded in the HEX A gene located on chromosome 15q 23-24 and the β subunit is encoded in the HEX B gene located on chromosome 5q 13.9 A defect of the β subunit leads to total absence of both β-hexosaminidase A and B, and gives rise to Sandhoff disease, while a defect of the α subunit results in Tay-sachs disease due to the absence of β-hexosaminidase A and S.1
Tay-sachs disease is an exclusively neurologic degenerative disorder manifesting the same ophthalmic symptoms found in Sandhoff disease, such as optic atrophy and a cherry red spot in the macula. It is caused by a deficiency of lysosomal hydrolase β-hexosaminidase A and β-hexosaminidase S causing accumulation of its substrate, GM2 ganglioside, in the lysosome of neuronal cells. This disease is transmitted by an autosomal recessive pattern and occurs among Ashkenazi Jews with an unusually high frequency, as the carrier frequency is about 1 in 25 Jewish birth. The clinical phenotype of Tay-sachs disease is essentially similar to that of Sandhoff disease, but typically there is no organomegaly or skeletal abnormality that can be seen in Sandhoff disease.11
Age of onset has been correlated with the amount of residual GM2 ganglioside degrading activity present in patient's cells.2 The infantile form of Sandhoff disease is characterized by an early onset of symptoms, which usually occur in the first 6 to 18 months of life with progressively severe neurologic impairment and generalized hypotonia. The infantile form is fatal leading to death before the age of 4 years.3 The peculiar cherry red spot in the macula is due to the accumulation of sphingolipid in retinal ganglion cells. Retinal ganglion cells, which have a substantial amount of sphingolipids, lose their transparency. In relation, the macula maintains its transparency because they are free of ganglion cells. As the disease progresses, optic atrophy can be present.3
When the disease invades the central nervous system, low signal intensity at the thalamus and high signal intensity at the basal ganglia and cerebral white matter are seen in T2-weighted brain MR images. These features are caused by the accumulation of calcium associated with the intracellular storage of GM2 ganglioside and are defined as characteristics of the infantile form of Sandhoff disease.12,13
This had a family history and showed severe neurologic impairment from the age of 12 months. A lysosomal enzyme assay of peripheral blood showed an absence of both β-hexosaminidase A and B isoenzymes in the serum. Further enzyme assay using fibroblasts showed a considerable reduction of both total β-hexosaminidase A and B. In particular, the percent of β-hexosaminidase A was elevated. β-hexosaminidase A is composed of one α subunit and β subunit, while β-hexosaminidase B is composed of two β subunits.10 An elevated percentage of β-hexosaminidase A compared to that of β-hexosaminidase B in the infantile form of Sandhoff disease can be explained by the excess α subunits, when fewer β subunits are made.1
T2-weighted brain MR images in this patient showed low signal intensity at the bilateral thalamus and high signal intensity at the cerebral white matter. There was a general atrophy in the cerebral cortex and cerebellum.
The treatment is symptomatic for the infantile form of Sandhoff disease, and generally involves management of the epileptic seizures and an intervention program for the motor and mental retardation. Case fatality in the infantile form typically occurs before the age of 4 due to extensive and severe central nervous deterioration.3 A poor prognosis is expected for this patient with regard to development and visual ability because she still showed severe neurologic impairment and no progression of ophthalmologic symptoms. In conclusion, we diagnosed this patient as having infantile form of Sandhoff disease showing bilateral optic atrophy and a cherry red spot in the macula through characteristic clinical features, neurologic image study, and laboratory findings.


 XML Download
XML Download