

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Cornelia de Lange syndrome was first reported by Cornelia de Lange, a Dutch pediatrician. Cornelia de Lange Syndrome includes developmental delay, growth retardation, low birth weight, skeletal formation anomaly, hirsutism and characteristic facial appearance.1 This syndrome is also called Brachmann de Lange syndrome since he reported a patient with similar symptoms in 1919. To date, around 800 cases have been reported internationally from diverse medical fields, but only a few cases have been reported in the ophthalmologic area. In Korea, since the first report of this syndrome by Moon et al. in 1967, several cases have been reported by pediatricians. In the 1990's, 2 cases of limb malformation such as syndactyly were reported, but there has been no case report of facial abnormalities or oculoplastic corrections. Therefore, we present a case of Cornelia de Lange syndrome in an 18-year-old female.
Case Report
An 18-year-old female presented with corneal irritation by her upper and lower cilia in both eyes.
She was born at term, but at low weight of 1.9 kg. She was diagnosed with ventricular septal defect and had an operation for her cleft palate at the age of 5. She attended a special school for having psychological lag and developmental impairment.
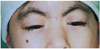
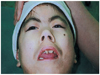
On her ophthalmological examinations, she showed bilateral symmetrical ptosis with IPF 5 mm and MRD1 1.5-2 mm and had no signs of Bell's phenomenon. Visual acuity was uncheckable because of her noncooperation but she showed orthophoria in the primary position. Besides, she had superficial keratitis caused by corneal irritation accompanied with long and dark curly cilia, cutaneous fistula of the nasolacrimal duct, and entropion. She had a large amount of hair, dark eyebrows, general hirsutism, a short glabella, mildly lifted nostrils, a protruded mouth, and a thin, drooped, lower lip. Her hands and feet were small and her distal interphalangeal joints were bent. Her secondary sexual characteristics showed late at the age of 18 and she only weighed 20 kg. Despite her youth, she showed early signs of aging such as white hairs.
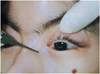
We repaired entropion by Hotz operation and electrocautery on both lower eyelids under general anesthesia. An examination under general anesthesia revealed high myopia in both eyes with right eye -7.00 diopters and left eye -6.00 diopters. There was also optic nerve atrophy, tigroid patterned myopic retinal change, and lacrimal cutaneous fistula in both eyes. Intraocular pressure was 8mmHg in both eyes by Tono-pen. Because she had lagophthalmos without Bell's phenomenon, no procedure was performed to repair the ptosis. We ordered glasses for her myopia. For the superficial keratitis, 0.3% ofloxacin, sodium hyaluronate eyedrop, erythromycin ointment were prescribed (Fig. 1, 2, 3, 4).
Discussion
The incidence of Cornelia de Lange syndrome is 1 per 10,000-100,000 live births.2-4 The etiology is still unknown but some theories have been proposed including dominant/recessive inheritance and chromosomal anomaly.5-9 In terms of genetics, although most cases of Cornelia de Lange Syndrome occur sporadically, the repetition of chromosome 3q26-27 is often seen in a number of cases. The systemic symptoms of Cornelia de Lange syndrome are shortness of stature of prenatal onset, mental retardation and sluggish physical activity, initial hypertonicity, microbrachycephaly, long, curly eyelashes, small nose, anteverted nostrils, characteristic lips and mouth, late eruption of widely spaced teeth, micrognathia, hirsutism, hypoplastic nipples and umbilicus, simian crease, proximal implantation of thumbs, micromelia, hypospadias, etc. Other less frequent findings are seizure attack (20%), cleft palate, congenital heart disease (29%, ventricular septal defect is the most common), hernia, and intestinal malrotation.1,10,11 Variable clinical manifestations have been reported by different studies on Cornelia de Lange syndrome.5,6,10,11,12 Examinations in little children are sometimes not possible because of their aggressive behavior, or photophobia without definite cause. Moreover, owing to their aggressive behavior, they are not willing to wear the prescribed glasses and unsuccessful occlusion therapy for amblyopia had also been reported. In addition, the ophthalmologic symptoms of Cornelia de Lange syndrome are not yet fully understood and forehead hirsutism and connected eye brows were present in 85-100% of the patients.3,11,13 In Alex V. Levin's study on 22 children suffering from Cornelia de Lange syndrome, 45% had ptosis, 36% had nystagmus, and 60% were in myopic condition, 28% of which had severe myopia over -5 diopters. In addition, the study reported that 82% were orthophoria, and that most of the children had no anterior segment anomaly. Also, no nasolacrimal duct anomaly was found except in a child with lacrimal fistula.13 On the other hand, Vila-Coro's14 study reported cases with obstruction of the nasolacrimal duct and many other studies showed esotropia, exotropia, and amblyopia in Cornelia de Lange syndrome. Corneal opacity, corectopia, microcornea, nanophthalmos, astigmatism, optic nerve atrophy, optic nerve coloboma, and exophthalmos have also been reported in a few cases.5,10,12,14,15,16
In this case study, the following findings of Cornelia de Lange syndrome were found: low birth weight, cleft palate, developmental delay, psychological lag, small hands and feet, skeletal anomaly such as bent distal interphalangeal joints, congenital heart disease such as ventricular septal defect, general hirsutism, delay in secondary sexual characteristics and early signs of aging.
The ophthalmologic symptoms of Cornelia de Lange syndrome include long curly cilia, ptosis, severe myopia, and cutaneous fistula of the nasolacrimal duct. The patient had the characteristic facial appearance of Cornelia de Lange syndrome including hirsutism, dark eye brows closely attached, lifted nostrils and a protruded mouth.
Children with Cornelia de Lange syndrome often suffer psychological lag and developmental delay as they are often diagnosed by pediatricians in the first place. For patients with Cornelia de Lange syndrome, careful examinations should be done on the variable symptoms listed above to minimize the possible development of complications and to improve the quality of life.

 XML Download
XML Download