

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Aicardi syndrome is a severe congenital disorder characterized by infantile spasms chorioretinal lacunae, and agenesis or hypogenesis of the corpus callosum. It was first identified by a French neurologist, Jean Aicardi in 1965.1,2 Multiple cranial, ocular, and skeletal malformations are main manifestations of the patients. It is an X-linked dominant disorder, thought to be secondary to a random, sporadic mutation. The syndrome is seen almost exclusively in females, since its early embryonic lethality is observed in the male patients.3
This article describes a 6 month old female patient with Aicardi syndrome who manifested complex partial seizure and myoclonic seizure. A magnetic resonance imaging(MRI) scan of the brain confirmed hypogenesis of corpus callosum, chorioretinal lacuane, and there were atypical EEG finding as well.
CASE REPORT
The female girl was born in November 2002, at full term by a vaginal delivery, after a healthy pregnancy. She was the first baby of a young mother, with history of essential hypertension. At 6 months of age, the baby developed abnormal eye movement and seizures of the complex partial type and a myoclonic type. The medical history of the patient included a gastroesophageal reflux at 2 months old.
On the physical examination, the head was normocephalic with a head circumference of 41cm(in the 10th percentile). Neurological examination revealed normal muscle tone.
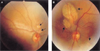
Ophthalmologic examination revealed no microphthlamia. Funduscopic examination revealed bilateral variable sized, discrete, two loci of pale areas with sharp borders, some of which had minimal surrounding pigmentation (chorioretinal lacunae), these were especially clustered around the disc, and were more prominent on the left eye(Fig. 1). The physical examination of other systems and the serological test for a TORCH group of infections were normal. An electroencephalography (EEG) of the patient showed abnormality. There was interhemispheric asymmetry with an epileptogenic discharge in the centroparietal area of both hemisphere and this was more prominent on the right side but there was no evidence of hypsarrhythmia.
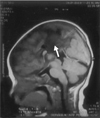
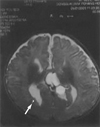
The brain MRI pictures of the patient revealed the presence of genu associated with agenesis of the rest of corpus callosum(Fig. 2). There was a multilobulated midline cystic mass with CSF intensity, which was lodged at interhemispheric area and there was a right subependymal gray matter heterotopia(Fig. 3). She was taking a antiepileptic drug and she showed good seizure control. On the latest follow up examniation, she had no abnormal eye movement and she showed good fixation and follow in both eyes.
DISCUSSION
Aicardi syndrome is characterized by the diagnostic triad of infantile spasms, chorioretinal lacunae, and an agenesis or hypogenesis of the corpus callosum.1,2 Aicardi syndrome is a rare X-linked dominant genetic disorder. The syndrome is seen almost exclusively in females, with early embryonic lethality in hemizygous males.4,5 The genetic studies for our patient was normal.
The age range of affected patients are from birth to the middle 20s. The patients with Aicardi syndrome are most commonly identified between the ages of 3 and 5 months. Most of the patients seem to develop normally until around the age of 3 months, when they begin to have the characteristic clinical findings associated with Aicardi syndrome.1,2,6-9
The ophthalmologic examination of the patients usually shows bilateral but sometiems, asymmetric ocular anomalies. Funduscopic examination shows pathognomonic chorioretinal lacunae; this was also clearly observed in our case, which are well defined, multifocal, pale areas with minimally pigmented borders, and they are usually clustered around the optic disc. The chorioretinal lacunae are the peculiar punched out areas of choroidal and RPE atrophy with foci of papillary proliferation of the RPE. These structure are observed mainly in the lacunar defect of the RPE and underlying atrophic choroid.10-15
Other possible ocular associations are microphalmia, optic disc or nerve coloboma, persistant hyperplastic primary viterous16 and so on. In our case, none of these findings were observed with the exception of the characteristic chorioretinal lacunae.
Aicardi syndrome should be considered as a syndrome in which the clinical findings and prognosis are heterogeneous. The patients present with earlyonset infantile spasms or spasm-like epileptic seizures, severe mental retardation, and severe limitation of motor development and language. The epileptic seizures are often difficult to control, and EEG studies may reveal a pattern characteristic of these spasms, called hypsarrhythmia. Our patient did not reveal infantile spasm and the electroencephalography( EEG) of our patient showed abnormal,
Interhemispheric asymmetry with an epileptogenic discharge in the centroparietal area of both hemispheres. This was more prominet on the right side but there was no evidence of hypsarrhythmia. Intracranial anomalies include callosal agenesis or hypogenesis. The corpus callosum normally develops from the anterior to posterior direction. In our case, hypogenesis of posterior corpus callosum with a normal anterior section, suggests that the corpus callosum initially developed and then it regressed. Since the exact nature of the corpus callosum malfomation in the syndrome remains unclear, the atypical nature of our patient's corpus callosum does not rule out the diagnosis of Aicardi syndrome. Callosal dysgenesis is typically associated with interhemispheric cysts and gray matter heterotopia, they are located mostly periventricular or subcortical area.11 These were also clearly observed in our case.
Costovertebral malformations such as hemivertebrae, fusion of vertebrae, kyphoscoliosis, absent or malformed ribs, and the occasionally cleft lip and palate may also be associated with Aicardi syndrome.8,17 None of these findings were detected in our case.
The spectrum of motor and mental disabilities is wide and, their severity is to some extent determined by the severity of underlying brain abnormalities. Some patient with milder brain malformations may have some understanding of the language, an independent motility, and they may be responsive to their enviornment.17-19 The treatment is symptomatic for Aicardi syndrome, and this generally involves management of the epileptic seizures and an intervention program for the motor and mental retardation. The prognosis of the patients varies with the severity of underlying brain abnormalities and symptoms, and the life expectancy can be severely limited from several months to only a few years.17 In our case, a favorable prognosis could be expected because the infantile spasms had not developed since birth. Also, a favorable prognosis is expected because the agenesis of corpus callosum is partial and the chorioretinal lacunae are relatively small in size, isolated and they spare the macula.20
In conclusion, we were presented with a 6-months-old female patient with Aicardi syndrome who manifested the unusual findings of a complex partial seizure, myoclonic seizure and atypical EEG finding in addtion to the well-known characteristic clinical and imaging findings of the syndrome. We have reported on this case along with a review of the literature.
 XML Download
XML Download