

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Waardenburg syndrome (WS) is a rare, autosomal dominant disorder characterized by sensorineural hearing loss, pigmentary disturbances of the skin, hair, and iris, and other developmental defects such as lateral displacement of both medial canthi and lacrimal puncta called dystopia canthorum.1 This syndrome is caused by the physical absence of melanocytes from the skin, hair, eyes, or stria vascularis of the cochlea. WS has variable penetrance and gene expression of clinical pictures.2
There are four types of this syndrome.3,4 WS types 1 and 2 can be distinguished clinically only by dystopia canthorum. Dystopia canthorum is the most penetrant feature of WS type 1, being present in 99% of those affected, but not in WS type 2.5 Apart from dystopia canthorum, all features of WS types 1 and 2 show marked interfamilial and intrafamilial variability. WS type 3 features musculoskeletal abnormalities. The presumed recessive combination of pigmentary disturbance and Hirschsprung's disease has been called Shah-WS, or WS type 4.
A few clinical descriptions of WS types 1 and 2 have been reported in Korea.6-8 Nowadays, WS is known to be caused by a gene alteration, mainly by mutations. While mutations of the PAX3 (paired box) gene have been identified in about 99% of WS type 1 cases, WS type 2 is a heterogeneous group, with about 15% of cases caused by mutations in microphthalmia associated transcription factor (MITF).9-11 However, MITF mutations are obviously not the major cause of WS type 2 and for most cases the genetic basis remains unknown.
We describe here three cases of WS type 2 in a Korean family and present the results of genetic studies.
CASE REPORTS
Case 1
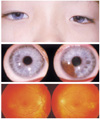
A 10-year-old boy (proband) was referred for ophthalmological evaluation because of iris heterochromia. It was intended that he would undergo artificial cochlear implantation in our hospital. He had congenital deaf-mutism. Best corrected visual acuity (BCVA) of both eyes was 1.0. Anterior segment examination was unremarkable except for iris heterochromia. His right eye showed complete iris hypopigmentation (pale blue eye), and his left eye showed iris hypopigmentation with segmental normal colored iris (Fig. 1). There was no evidence of dystopia canthorum. Fundus examination demonstrated generalized decrease in retinal pigment with a focal hypopigmented lesion in both eyes. No other abnormalities, such as hair or skin hypopigmentation, were found. Conventional audiological examinations showed profound, bilateral, sensorineural hearing loss (Fig. 2). In the genomic DNA sample of this patient, no abnormally migrating bands were seen in the PAX3 or MITF genes.
Case 2
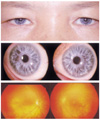
A 48-year-old mother of the proband had bilateral sensorineural hearing loss. Her BCVA was 1.0 in both eyes. Anterior segment examination was unremarkable with the exception of bilateral iris isohypochromia. Both her eyes showed complete iris hypopigmentation (pale blue eyes) (Fig. 3). There was no evidence of dystopia canthorum. Fundus examination demonstrated generalized decrease in retinal pigment with an area of hypopigmentation on the posterior pole in both eyes. The genetic studies revealed no mutation in either PAX3 or MITF genes.
Case 3
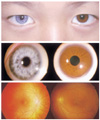
A 9-year-old brother of the proband also had sensorineural hearing loss. His BCVA was 1.0 in both eyes. Anterior segment examination was normal except for iris heterochromia. He had a blue-colored iris in his right eye and a dark brown-colored iris in his left eye (Fig. 4). There was no evidence of dystopia canthorum. Fundus examination demonstrated generalized decrease in retinal pigment with a focal hypopigmented lesion in his right eye and no pigmentary disturbances in his left eye. Hypopigmentation of the skin or hair was not found. The genetic studies revealed no mutation in either PAX3 or MITF genes.
A 8-year-old sister of the proband had no clinical features of WS (Fig. 5). Her ophthalmological and audiological evaluations were within normal limits.
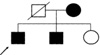
A family pedigree was constructed for this family (Fig. 6). The correct evaluation of the family's history of pedigree was difficult in this family because the family members did not live together and the three children inhabited different social welfare communities for the deaf. Their father had died several years ago. Based on the subjects' memory, their father had no features of WS.
DISCUSSION
WS is a dominantly inherited example of auditory pigmentary syndrome with patchy depigmentation of the skin, hair, eyes or stria vascularis of the cochlea. The diagnosis in patients with classic features of WS or in families with affected relatives is not difficult. Both sexes and all races are equally affected by WS. Unfortunately, not every case expresses all clinical manifestations of complete WS and the incomplete form is commonly described. The expression of clinical findings is extremely variable. WS type 1 is characterized by evidence of dystopia canthorum and the full disease symptomatology. The reported incidence of dystopia canthorum in patients with WS has ranged from 41.2% to 99%.3,12 Congenital deafness is clinically the most serious symptom. The deafness in WS type 2 individuals was found to be more common, more severe and more likely to feature the bilateral form. Hearing impairment can be explained by a lack of melanocytes in the stria vascularis of the cochlea. Cutaneous color abnormalities include achromic spots and hyperpigmented macules and are observed in 8.3% to 50% of patients.3,12 There are two types of pigmentary disturbances of hair in WS: white forelock and premature graying of scalp hair and of the eyebrows, cilia, or body hair (poliosis). White forelock is observed in 17% to 58.4% of cases, and premature graying in 7% of cases.3,12 Ocular color abnormalities of WS include three types of disturbances: iris heterochromia, bilateral iris isohypochromia, and fundus pigmentary alterations (generalized decrease in retinal pigment or pigmentary mottling in the periphery). Iris heterochromia, partial or complete, was found in 21% to 28% of patients with WS. Bilateral iris isohypochromia in WS was reported to occur in 14.9% to 42% of cases.3,12
WS type 2 is a heterogeneous group with normally located canthi (without dystopia canthorum). Sensorineural hearing loss (77%) and iris heterochromia (47%) are the most important diagnostic indicators for WS type 2.13 Both are more common in type 2 than in type 1. Other clinical manifestations, such as white forelock and skin patches, are more frequent in type 1.15
WS is caused by a gene alteration, mainly by mutations. Inherited causes account for about 50% of individuals seen for congenital or early childhood hearing loss, of which 70% are due to mutation in numerous single genes which impair auditory function alone.16 The remainder are associated with other developmental anomalies, and are termed syndromic deafness. Genes responsible for syndromic forms of hearing loss in WS include the PAX3 and MITF genes. WS is autosomal dominant for most cases with WS types 1, 2, and 3, but is autosomal recessive for WS type 4. Recently, mutational changes in the PAX3 gene in patients with WS type 1 have been identified.15 WS type 2 is a heterogeneous group, about 15% of which are heterozygous for mutations in the MITF gene.16 Whereas WS type 1 defines a specific genetic entity, the clinical definition of WS type 2 is arbitrary, covering any auditory-pigmentary syndrome that does not clearly belong somewhere else. All these forms show marked variability even within the same family.
In conclusion, we have presented a Korean, WS type 2 family in which no mutations of the PAX3 or MITF genes could be found. The genetic basis, as yet unknown for most cases of WS type 2, might be found with further investigation.

 XML Download
XML Download