

 PDF
PDF ePub
ePub Citation
Citation Print
Print



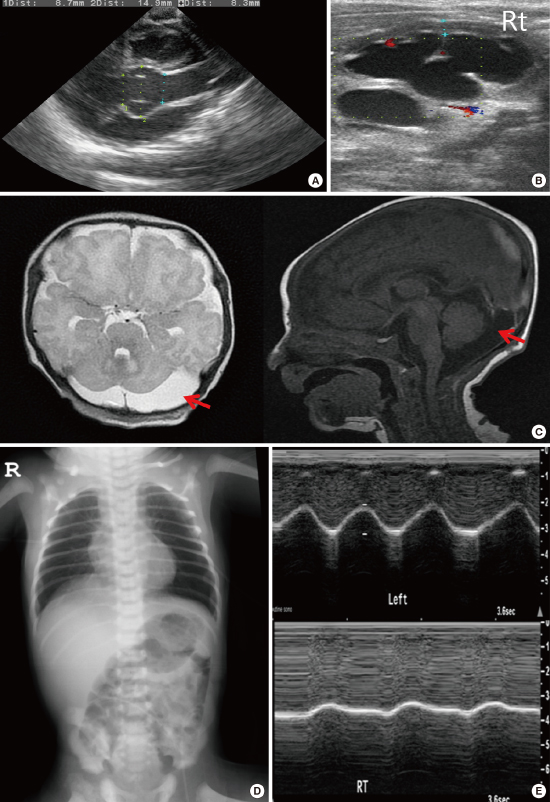
In July 2015, a male newborn was born vaginally at 36+5 weeks gestation to a G1P0 mother. The birth weight was 3,000 g (75th to 90th percentile), birth length was 54.5 cm (> 99th percentile), and head circumference was 35.5 cm (99th percentile). Physical examination revealed several dysmorphic features, such as facial dysmorphism with dolichocephaly, micro- and retrognathia, downslanting palpebral fissures, crumpled ears, marked arachnodactyly, an adducted thumb, reduced elbow extension and pectus excavatum. In ophthalmological investigation, no lens dislocation was noted. Echocardiography showed a dilated aortic root (aortic sinus diameter 14.9 mm, z-score 4.5) (Fig. 1A), mild aortic regurgitation, and redundant mitral valve chordae apparatus. The abdominal ultrasonography revealed grade 4 hydronephrosis and severe hydroureter in the right kidney (Fig. 1B). Brain magnetic resonance imaging confirmed a dilated cisterna magna, particularly on the left (Fig. 1C). In spine ultrasonography, no dural ectasia was noted. Chest radiography showed eventration of the right hemidiaphragm (Fig. 1D). Weak diaphragmatic motion and shallow excursion during respiration in the right hemidiaphragm were confirmed by dynamic thoracic ultrasonography (Fig. 1E).The systemic score was 6 points. Direct sequencing was performed for a total of 65 exons of the fibrillin-1 (FBN1) gene. The analysis revealed missense mutation at nucleotide 3217 (c.3217G>A) in exon 26 of the FBN1 gene, resulting in the substitution of a glutamate for a lysine at codon 1073 (p.Glu1073Lys) in the 12th calcium binding epidermal growth factor-like domain of the FBN1 protein. At 6 months of age, abrupt cardiac arrest caused by rupture of mitral chordae occurred, and surgical repair was conducted. He was prescribed captopril and is on follow-up. There were no abnormally tall relatives in the family and no family history of cardiac, ophthalmic, or genetic disease.
Marfan syndrome (MFS, OMIM: 154700) is a disorder of the connective tissue with autosomal dominant inheritance and is caused by mutations in the gene coding for FBN1, located on chromosome 15q21.1 (1). Significant phenotypic variability of MFS is commonly observed and neonatal MFS (nMFS) is the severe end of clinical spectrum of MFS, with death often occurring within the first 2 years of life due to congestive heart failure (234). And this severe, early-lethal manifestation has been correlated with mutations in exons 24–32 (3). The common features of nMFS include ascending aortic dilatation, severe mitral and/or tricuspid valve insufficiency, ectopia lentis, arachnodactyly, crumpled ear, and pulmonary emphysema (45). To our knowledge, there have been 2 nMFS cases to date in the literature with identical mutations to the present case (67). All the cases, including the present case, had severe cardiovascular abnormalities and common features like congenital contracture, arachnodactyly and crumpled ear. However, there were no patients other than the present case who had several atypical manifestations, such as diaphragmatic eventration, severe hydronephrosis with hydroureter, and dilated cisterna magna. Our report is the first atypical nMFS case with p.Glu1073Lys mutation of FBN1 in Korea and may help clinicians with the diagnosis and follow-up of atypical nMFS.
 XML Download
XML Download