

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Pachydermoperiostosis (PDP), or primary hypertrophic osteoarthropathy, is a cluster of genetic syndromes characterized by three major symptoms; digital clubbing, periostosis, and pachydermia. Additional manifestations including excessive sweating, acne, arthropathy, and acro-osteolysis of long bones have been reported (1). The phenotypic spectrum of PDP is broad, and three clinical presentations of PDP are generally recognized: the complete form involving all three major symptoms, the incomplete form with periostosis without pachydermia, and the ‘forme fruste’ with pachydermia and minimal or no skeletal anomalies (2).
Both autosomal dominant with incomplete penetrance and recessive inheritance have been suggested in PDP (3). The precise incidence and prevalence are still unknown. Uppal et al. (4) recently discovered homozygous mutations in the hydroxyprostaglandin dehydrogenase (HPGD) gene in a subset of patients with PDP. In addition, exome analysis of PDP in Japanese, Chinese, Caucasian, and other races has revealed homozygous and compound heterozygous mutations in the solute carrier organic anion transporter family member 2A1 (SLCO2A1) gene (5678). Both HPGD and SLCO2A1 genes are involved in prostaglandin E2 (PGE2) degradation. Increased levels of PGE2 in patients with causal genetic mutations could contribute to the pathogenesis of PDP (9).
To date, one family of PDP related to SLCO2A1 gene mutation has been reported in Korea (10). In this study, we describe complete types of PDP carrying SLCO2A1 mutations including a novel mutation in Koreans with review of literature.
MATERIALS AND METHODS
Subjects
All participants were of Korean ethnicity. Six patients with PDP and their family members from three unrelated families were recruited.
Family 1
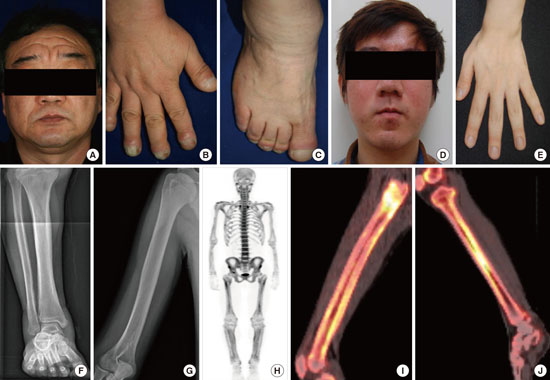
The case of this family (Fig. 1 and 2A-C) has been previously reported (11). The 56-year-old proband, who was born to healthy non-consanguineous parents, visited the clinic for genetic counseling of his son. At 19 years of age, the proband presented with enlarged hands and feet, knee joints swelling and pain and was then referred to a neurosurgeon for suspicion of acromegaly. He underwent hypophysectomy, which showed no tumor lesion in the specimen. Attention was aroused only after periosteal thickening was observed in the skeletal survey of the hands and forearm bones, indicating primary hypertrophic osteoarthropathy as well as facial furrowing. His two brothers, who were also diagnosed as PDP, showed similar clinical features to the proband.
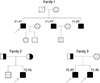 | Fig. 1Pedigrees of affected individuals with SLCO2A1 mutations. In the pedigree, arrows indicate the proband. Filled black, patients with PDP; Half-black, healthy members with a heterozygous mutation; Gray, healthy members not doing genotyping test.
|
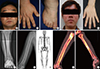 | Fig. 2Clinical features of affected individuals with SLCO2A1 mutations.
(A-C) Clinical pictures of family 1 proband. (A) Thickening and furrowing of the facial skin. (B-C) Digital clubbing and swelling of the ankle joint. (D-H) Clinical pictures of family 2 proband. (D) Thickening and greasiness of facial skin. (E) Digital clubbing. (F-G) Cortical hyperostosis of long bones. (H) Diffusely increased uptake in the whole axial and appendicular long bones shown by whole body bone scan. (I-J) 18F-fluoride PET scan images of femur and tibia show increased cortical/periosteal uptake in the proband of family 3.
|
Family 2
The 19-year-old proband (Fig. 1 and 2D-H) was born to a healthy non-consanguineous couple. Beginning at 17 years of age, he noticed inappropriately thickened lower legs and swelling of ankle joints. He also complained of hyperhidrosis, acne, and difficulty of squatting. He had atrial septal defect (ASD), patent ductus arteriosus (PDA), and valvular heart disease and underwent an operation for PDA at the age of 1 month. A physical examination revealed enlargement of the terminal phalanges and facial furrowing. Skeletal survey showed periosteal thickening of the long bones. The radionuclide whole body bone scan (WBBS) showed diffusely increased uptake in the whole axial and appendicular long bones, suspicious of metabolic bone disease. Results of laboratory analyses, including growth hormone, insulin-like growth factor 1, and thyroid-stimulating hormone were normal. His parents were not clinically affected.
Family 3
The case of this family was recently reported during preparation of this manuscript (10). Two siblings (Fig. 1) of ages 23 and 19 years presented with progressive pachydermia and enlargement of their hands and feet since puberty. They also complained of hyperhidrosis, acne, and frontal furrowing. The brothers suffered from watery diarrhea; however, colonoscopic finding was not remarkable. The younger patient had a history of the operation for PDA in infancy; whereas his older brother had no history of PDA or other congenital heart diseases. Skeletal survey showed periosteal thickening of the long bones. The 18F-fluoride positron emission tomography (PET) scan showed increased cortical/periosteal uptake of long bones (Fig. 2I and 2J). Laboratory tests including growth hormone and insulin-like growth factor 1 were within normal limits. Their parents were healthy without any similar features.
Mutation analysis of the associated genes
A mutation analysis of the affected individuals and their family members was performed by direct sequencing of PCR products amplified from genomic DNA. Genomic DNA was extracted from peripheral blood leukocytes using conventional methods. The DNA sequences of SLCO2A1 and HPGD were obtained from the available online database (NM_005630.2 and NM_000860.4, respectively). Primers were designed using Primer 3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/). All exons and their exon-intron boundaries in SLCO2A1 and HPGD were amplified via PCR. Direct sequencing was performed using the BigDyeTM Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Waltham, MA, USA).
RESULTS
Clinical features of PDP patients
Based on major diagnostic criteria (digital clubbing, periostosis, and pachydermia) (1), all patients showed major clinical findings and conformed to the diagnosis of PDP. They were categorized as having the complete form and developed major symptoms before the age of 20 years and subsequently suffered from progressive skin thickening with seborrhea, swelling of periarticular tissue, and joint discomfort or arthralgia. Their parents did not show clinical signs of PDP. The clinical phenotypes of PDP patients are summarized in Table 1.
Table 1
Clinical phenotypes of Korean patients with pachydermoperisostasis

| Phenotypes | Cases | |||||
|---|---|---|---|---|---|---|
| F1-P1 | F1-P2 | F1-P3 | F2-P4 |
F3-P5 Kim et al. (10) |
F3-P6 Kim et al. (10) |
|
| Current age, yr | 56 | 54 | 52 | 19 | 23 | 19 |
| Onset age, yr | 19 | 17 | 20 | 17 | puberty | 13 |
| Gender | M | M | M | M | M | M |
| SLCO2A1 mutations | c.302T>G | c.302T>G | c.302T>G | c.940+1G>A | c.940+1G>A | c.940+1G>A |
| c.1807C>T | c.940+1G>A | c.940+1G>A | ||||
| Triad | ||||||
| Digital clubbing | + | + | + | + | + | + |
| Periostosis | + | + | + | + | + | + |
| Pachydermia | + | + | + | + | + | + |
| Skin | ||||||
| Palmar and plantar hyperhidrosis | + | - | - | - | - | - |
| Acne | + | + | + | + | + | + |
| Seborrhoea and eczema | + | + | + | + | + | + |
| Skeletal | ||||||
| History of bone fractures | - | - | - | - | - | - |
| Swelling of large joints | + | + | + | + | + | + |
| Painful joints on exercise | + | + | + | + | + | + |
| Hydrarthrosis | + | + | + | + | + | + |
| Others | ||||||
| Anemia | - | - | - | - | - | - |
| Hypoalbuminemia | - | - | - | - | - | - |
| Patent ductus arteriosus | - | - | - | + | - | + |
![]()
Identification of SLCO2A1 as a gene responsible for PDP
PCR was performed for screening of SLCO2A1 and HPGD mutations followed by a direct sequence analysis, and no HPGD mutation was found in affected individuals. The results of these genetic analyses are shown in Fig. 3.
 | Fig. 3Localization and sequence chromatogram of identified SLCO2A1 mutations. Upper: The positions of the mutations in the exons of SLCO2A1 in this study. Lower: SLCO2A1 mutations in PDP families. family 1, (A) c.302T>G; family 2, (B) c.940+1G>A and (C) c.1807C>T; family 3, (C) c.940+1G>A.
|
In family 1, DNA sequencing of the proband revealed a novel heterozygous mutation in the SLCO2A1 gene at nucleotide 302 (c.302T>G) causing a substitution of the amino acid isoleucine (Ile) to serine (Ser) at codon 101 (p.Ile101Ser) in exon 3 as well as a known polymorphism (A396T) in exon 14. The two brothers of the proband were also heterozygous for c.302T>G. The proband’s son, who seemed normal in appearance, did not carry the mutations.
In family 2, the proband showed compound heterozygous for 2 different SLCO2A1 mutations; c.940+1G>A and c.1807C>T. The c.940+1G>A mutation was located in the splice donor site of intron 7, which resulted in the loss of exon 7 and a truncation of prostaglandin transporter (512), and c.1807C>T in exon 13 introduced a stop codon at position 603 (p.Arg603*). The patients’ father and mother were heterozygous for c.940+1G>A and c.1807C>T, respectively.
In family 3, the brothers were homozygous for c.940+1G>A. The patients’ father was heterozygous for c.940+1G>A, and evaluation of the mother was not available.
DISCUSSION
PDP or primary hypertrophic osteoarthropathy is characterized by pachydermia, periostosis and finger clubbing; however, the phenotypic spectrum of PDP is known to be broad (1). The diagnosis of PDP is based on the typical clinical features and radiological data. PDP needs to be differentiated from acromegaly and secondary hypertrophic osteoarthropathy that is associated with pulmonary or cardiac disease. For the differential diagnosis, hormone tests including growth hormone and thyroid stimulating hormone, absence or presence of thickening and furrowing of facial skin, and sella turcica imaging might be helpful (13).
Since PDP was first described in 1868 (1), mutations in HPGD and SLCO2A1 have been identified in families affected with PDP (456). So far, 43 different SLCO2A1 mutations have been reported and are summarized in Table 2. Our study identified a novel mutation, c.302T>G, as well as known mutations, c.940+1G>A and c.1807C>T, in the SLCO2A1 gene. The c.940+1G>A mutation was suggested as a hot spot in both Chinese Han and European Caucasian patients with PDP (3614). Sasaki et al. (5) insisted that the c.940+1G>A mutation is a major mutation as an ancient founder allele in Japanese PDP patient. Considering ethnic roots and regional proximity, the c.940+1G>A mutation might also be a common mutation detected in Korean PDP patients.
Table 2
Summary of the genetic SLCO2A1 mutations in patients with pachydermoperisostasis in the literature
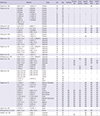
| References | Mutation | Origin | Sex | Age | Clubbing | Periostosis | Pachydermia | Hyperhidrosis | Seborrhea | Arthralgia | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Zhang et al. (19) | c.940+1G>A | c.1602C>A | Chinese | M | 22 | + | + | + | + | NA | + |
| Zhang et al. (3) | c.855delA | c.855delA | Chinese | M | 36 | + | + | + | - | + | + |
| c.855delA | c.855delA | Chinese | F | 47 | - | - | - | - | - | - | |
| c.855delA | c.855delA | Chinese | F | 42 | - | - | - | - | - | - | |
| c.1106G>A | c.1106G>A | Chinese | M | 23 | + | + | + | + | + | + | |
| c.1393G>A | c.1393G>A | Chinese | M | 26 | + | + | + | - | + | + | |
| c.493G>T | c.1136G>A | Chinese | M | 18 | + | + | + | - | + | + | |
| c.664G>A | c.1634delA | Chinese | M | 24 | + | + | + | - | + | + | |
| c.861+2T>C | Chinese | M | 42 | + | + | + | - | + | + | ||
| c.1065dupA | Chinese | M | 17 | + | + | + | + | + | + | ||
| Zhang et al. (31) | c.235-1G>T | c.656C>T | Chinese | M | 27 | + | + | + | + | NA | + |
| Zhang et al. (6) | c.97-1G>A | c.97-1G>A | Chinese | M | 24 | + | + | + | NA | NA | NA |
| c.764G>A | c.1634delA | Chinese | M | 27 | + | + | + | NA | NA | NA | |
| c.664G>A | c.940+1G>A | Chinese | M | 21 | + | + | + | NA | NA | NA | |
| Cheng et al. (32) | c.547G>A | c.1807C>T | Chinese | M | 25 | + | + | + | + | + | + |
| c.940+1G>A | c.1602C>A | Chinese | M | 37 | + | + | + | NA | + | + | |
| Niizeki et al. (12) | c.940+1G>A | c.1279_1290del12 | Japanese | M | 19 | + | + | + | + | + | - |
| c.754C>T | c.1807C>T | Japanese | M | 21 | + | + | + | + | - | + | |
| c.421G>T | c.940+1G>A | Japanese | M | 20 | + | + | + | + | + | - | |
| c.940+1G>A | c.1807C>T | Japanese | M | 20 | + | + | + | - | + | + | |
| Sasaki et al. (5) | c.940+1G>A | c.1279_1290del12 | Japanese | M | 24 | + | + | + | + | + | + |
| c.310G>A | c.1040C>T | Japanese | M | 25 | + | + | + | + | + | + | |
| c.940+1G>A | c.940+1G>A | Japanese | M | 45 | + | + | + | - | + | - | |
| c.940+1G>A | c.1668G>C | Japanese | M | 53 | + | + | + | - | - | + | |
| Niizeki et al. (9) | c.1279G>A | c.1807C>T | Japanese | F | 67 | + | + | - | - | - | + |
| Minakawa et al. (33) | c.940+1G>A | c.1279_1290del12 | Japanese | M | 15 | + | + | + | + | + | + |
| Busch et al. (14) | c.940+1G>A | c.1668G>C | Japanese | M | 53 | + | + | NA | NA | NA | + |
| c.940+1G>A | c.940+1G>A | Japanese | M | 21 | + | NA | + | NA | NA | NA | |
| c.940+1G>A | c.940+1G>A | Japanese | M | 19 | + | NA | + | NA | NA | NA | |
| c.1292delC | c.1292delC | Indian | M | 27 | + | NA | + | NA | + | + | |
| c.763G>A | c.763G>A | Indian | M | 26 | + | + | NA | + | NA | + | |
| c.763G>A | c.763G>A | Indian | M | 28 | + | + | NA | + | NA | + | |
| Seifert et al. (8) | c.830_831insT | c.830_831insT | Turkish | M | 21 | + | + | + | + | + | + |
| c.830_831insT | c.830_831insT | Turkish | M | 19 | + | + | + | + | + | + | |
| c.830_831insT | c.830_831insT | Turkish | M | 7 | - | - | - | - | - | - | |
| c.830_831insT | Turkish | M | 40 | + | - | - | - | - | - | ||
| c.1670T>C | c.1670T>C | Iraq | M | 38 | + | + | + | + | + | + | |
| c.754C>T | Dutch | M | 28 | + | - | - | - | - | - | ||
| Diggle et al. (7) | c.1259G>T | c.1259G>T | Hispanic (Colombia) | M | 45 | + | + | + | NA | NA | NA |
| c.941-1G>A | c.1517C>A | Chinese | M | NA | + | + | + | NA | NA | NA | |
| c.542G>C | c.542G>C | Turkish | M | NA | + | + | + | NA | NA | NA | |
| c.1333C>T | Dutch | M | NA | + | + | + | NA | NA | NA | ||
| c.290G>A | c.940+2T>A | French | M | NA | NA | NA | NA | NA | NA | NA | |
| c.664G>A | c.664G>A | North African | M | NA | NA | NA | NA | NA | NA | NA | |
| c.253A>T | c.253A>T | North African | M | NA | NA | NA | NA | NA | NA | NA | |
| c.1105+4A>G | c.1105+4A>G | Dutch | M | NA | NA | NA | NA | NA | NA | NA | |
| c.838C>T | c.1693T>G | Kabardin (Caucasus) | M | NA | NA | NA | NA | NA | NA | NA | |
| c.310G>T | c.310G>T | Italian | M | NA | NA | NA | NA | NA | NA | NA | |
| c.724+1G>T | c.724+1G>T | Algerian | M | NA | NA | NA | NA | NA | NA | NA | |
| c.542G>A | c.542G>A | Turkish | M | NA | NA | NA | NA | NA | NA | NA | |
| c.611C>T | c.611C>T | Italian | M | NA | NA | NA | NA | NA | NA | NA | |
| Ayoub et al. (34) | c.1016C>T | c.1016C>T | Saudi | M | 23 | + | + | + | + | + | + |
| Madruga Dias et al. (35) | c.940+1G>A | c.940+1G>A | African | M | 26 | + | + | + | NA | NA | + |
| Saadeh et al. (36) | c.838C>T | c.838C>T | Lebanese | M | 22 | + | + | + | NA | NA | + |
| c.838C>T | c.838C>T | Lebanese | M | 24 | + | NA | + | NA | NA | + | |
![]()
A considerable number of PDP patients showed autosomal recessive transmission. However, autosomal dominant transmission with incomplete penetrance has been also reported in previous studies (3). In this study, three patients of family 1 carried a heterozygous c.302T>G mutation. The mutated alleles supposed to be autosomal dominant inheritance were c.302T>G, c.754C>T, c.830_831insT, c.861+2T>G, c.1065dupA, and c.1333C>T (378). The patients having c.754C>T and c.830_831insT alleles showed mild clinical symptoms, whereas PDP patients carrying c.302T>G, c.861+2T>G, c.1065dupA, and c.1333C>T alleles presented the full-blown phenotype (Table 2). We found no consistent clinical features or genetically unique characteristics among those mutations.
Males were more commonly and severely affected with the ratio of male:female of 9:1 (13). A few females carrying the SLCO2A1 mutation were not clinically affected or showed atypical phenotype of PDP (379). A simple explanation of the skewed sex ratio is lacking. Plausible assumptions were testosterone promoting disease expression, weaker reactivity to prostaglandin in women than men, and suppressed effects of estrogen on interleukin 1b-mediated induction of the COX-2 pathway in vessels (11516).
Besides typical skin and bone manifestations, accompanying PDA was reported in about one third of cases in PDP patients with HPGD mutation (17). However, congenital cardiac anomaly has scarcely been reported in SLCO2A1-mutated patients. Only one Dutch patient carrying c.1333C>T mutation in the SLCO2A1 gene was reported to have ASD (7). Prostaglandin transporter–deficient mice by targeted deletion of the SLCO2A1 gene die as neonates probably as a result of a closure defect of the ductus arteriosus (18). Seifert et al. (8) reported that PDP patients with SLCO2A1 mutations did not show failure of postnatal ductus arteriosus closure. Contrary to previous reports, our patient carrying compound SLCO2A1 heterozygous mutations was diagnosed with congenital heart defects including ASD, PDA, and valvular heart disease. It is uncertain whether this case is a coincidental finding; or PDA might be a rare, fatal condition in SLCO2A1-mutated PDP patients.
Both HPGD and SLCO2A1 genes are known to involve PGE2 metabolism. Mutations in SLCO2A1 cause increased levels of PGE2 due to the failure of extracellular uptake of PGE2 (1920). Urinary excretion of the main PGE2 metabolite, PGE-M, is the best measure of total endogenous PGE2 production in normal condition (21). Previous studies showed elevated urinary excretion of PGE2 and PGE-M in PDP patients, suggesting that measurement of urinary PGE2 and PGE-M can be helpful in early and differential diagnosis of PDP (3412). Urinary PGE2 and PGE-M levels were not determined in the current study and further measurement is needed.
Increased prostaglandins, which have a biphasic role in bone metabolism, stimulate both bone resorption and formation (22). An increase in bone turnover markers was observed in young PDP patients who might have high disease activity (2324). Previously, bone scintigraphy confirmed a high activity of bone turnover in the affected joints of PDP patients (25). Three-phase skeletal scintigraphy in a PDP patient detected increased tracer uptake around long bones, which indicated the active form of the disease (26). Our patient in family 2 also showed similar finding of increased radionuclide uptake along the long bone cortices by WBBS (Fig. 2H). In this study, 18F-fluoride PET scan was performed in PDP patients for the first time. PET scan also provided convincing evidence of hypermetabolic status in various long bones.
There is no specific treatment for PDP (27). Clubbing is usually asymptomatic and does not require treatment (13). For patients with painful osteoarthropathy, the therapeutic options consist of salicylates, non-steroidal anti-inflammatory drugs, systemic corticosteroids and colchicine (27). Bisphosphonates or infliximab have been attempted in cases of PDP refractory to conventional therapy (2829). Cosmetic surgery or botulinum toxin injections have been introduced for correcting rough facial features (1330).
We report on three unrelated PDP families and identified three different mutations in the SLCO2A1 gene, of which c.302T>G in exon 3 is novel. As with Japanese and Chinese studies, we determine that c.940+1G>A is a major mutation in Korean PDP patients. Although our study did not contain PG-related metabolic activity, we further support the role of mutations in the SLCO2A1 gene in the pathogenesis of PDP.
 XML Download
XML Download