

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Physiological level of glucose stimulates insulin secretion; however, chronic hyperglycemia causes insulin secretory dysfunction, known as glucotoxicity (1).
Fatty acids (FAs) are also important for insulin secretion; however, prolonged exposure to elevated levels of FAs causes pancreatic β-cell dysfunction and apoptosis, resulting in lipotoxicity, which is augmented by high glucose (2,3).
Cellular uptake of fatty acids involves two components of passive diffusion through the lipid bilayer and protein-facilitated transfer playing a major role in metabolic tissues. Several proteins, including fatty acid translocase/cluster determinant 36 (CD36), family of fatty acid transport proteins (FATPs) (1-6), and plasma-membrane-associated FA-binding protein (FABPpm) have been identified as active FAs transport proteins.
CD36 has been documented having an important role of FA transport, and it is mainly distributed in liver and muscle tissues. Recently it was reported that CD36 is also expressed in insulin producing cells including MIN-6 cell, INS-cell, and human β-cell (4).
A previous report demonstrated that high glucose stimulated CD36 expression and palmitate uptake in INS-1 cells along with decreased insulin secretion, which was normalized by suppression of CD36 (5).
Intestinal absorption of lipid is regulated by various transporters, including CD36, scavenger receptor class B type I (SR-BI), and Niemann-Pick C1-Like 1 (NPC1L1).
Ezetimibe is a lipid-lowering agent that blocks Niemann-Pick C1-Like 1 (NPC1L1)–mediated cholesterol absorption in the apical brush border membrane of jejuna enterocytes. Following oral administration, over 80% of ezetimibe is rapidly absorbed and metabolized within the intestinal mucosa to form active ezetimibe-glucuronides, which are then transported through the portal vessel to the liver, where they undergo further glucuronidation and subsequent biliary secretion into the intestine (6). Binding of ezetimibe to CD13 in the brush border membrane of the small intestine has been reported to block cholesterol uptake (7) and subsequent signaling through CD13 requires co-association with additional proteins, such as CD36, CD64, or CD16. Ezetimibe induces conformational change in the extracellular domain of CD13 with masking of the co-assembly with these co-receptor proteins (8). In addition, ezetimibe was reported to induce a significant decrease in surface expression of CD13, CD16, CD64, and CD36 of monocyte-derived macrophages (9). Ezetimibe is also known to improve glucose metabolism in addition to lowering lipid levels in type 2 diabetes (10,11). Ezetimibe could inhibit the activity of hepatic NPC1Ll, promoting the insulin signaling pathway, and then improve insulin resistance in the liver (12). Several studies with diabetic animal models have revealed that ezetimibe could improve first phase insulin secretion and protect β-cells (13,14), however, the specific mechanism is still unclear.
This study attempted to clarify the effects of ezetimibe on pancreatic secreting beta-cells exposed to high glucose and to determine whether these effects are related to change of CD36 expression.
MATERIALS AND METHODS
INS-1 cell culture
INS-1 cells were grown in 5% CO2 with 95% air at 37°C in RPMI-1640 medium (GIBCO, Grand Island, NY, USA) containing 11.1 mM pyruvate, 10 mM HEPES, 50 M 2-mercaptoethanol, 100 U penicillin/mL, and 100 g streptomycin/mL. The RPMI-1640 medium used in all experiments contained the supplements described above. The cells were passaged weekly after they had been detached with trypsin–EDTA. All experiments were performed using INS-1 cells between passages 21 and 29.
Pancreatic islet isolation and cell culture
Pancreases from male Sprague-Dawley rats weighing 180-250 grams were infused with 10 mL of 1.5 mg/mL collagenase type XI (Sigma, St. Louis, MO, USA)/1% fetal bovine serum/2 units/mL RQ1 DNase (Promega, Madison, WI, USA.) solution in Medium 199 (Sigma). After surgical excision of the pancreas, it was incubated in collagenase solution at 37°C. Undigested tissue was removed using a 500 µm screen, and the recovered tissue was washed twice with ice-cold Hanks' balanced salt solution containing 0.1% bovine serum albumin, followed by centrifugation at 250 × g for 4 minutes. Prior to experimentation, islets in the pellet were separated using a Histopaque-1077 gradient (Sigma), and islets were then hand-picked and cultured in RPMI medium 1640 containing 10% fetal bovine serum, 11.1 mM glucose, and penicillin/streptomycin/amphotericin B.
mRNA expression of insulin and CD36 and intracellular peroxide levels were determined in INS-1 cells and primary rat islet cells exposed under normal (5.6 mM for INS-1 cells, 11 mM for primary cells) or high glucose condition (30 mM for both) with or without 20 μM of ezetimibe for 12 hours. The dose of ezetimibe was determined according to previous studies with monocyte or CaCO2 cells (7,9). Glucotoxicity was induced by three-day treatment with high glucose (30 mM) and glucose stimulated insulin secretion (GSIS) in INS-1 cells was evaluated. INS-1 cells were exposed to high dose palmitate (1 mM) for 24 hours.
Evaluation of reactive oxygen species (ROS) with flow cytometry
Intracellular peroxide levels were detected by flow cytometric analysis using an oxidation-sensitive fluorescein labeled dye, carboxylated dichlorodi-hydrofluorescein diacetate (carboxy-H2DCFDA, Molecular Probes, Carlsbad, CA, USA) (15). Upon oxidation by intracellular ROS, the non-fluorescent dye is converted into its fluorescent form. INS-1 cells were labeled with 100 M carboxy-H2DCFDA for 1 hour at 37°C. Following cell loading of the dye, the cells were washed twice with PBS and then put back into culture conditions for 2 hours. INS-1 cells were then harvested, washed twice with PBS, and re-suspended in trypsin–EDTA (0.25% trypsin, 2 mM Na4-EDTA, Invitrogen) for 5 minutes at 37°C. In order to disperse the cells into a single cell suspension, INS-1 cells were gently passed 20 times in and out of a 200–1,000 lL tip. The cells were then washed twice with ice-cold PBS. Analysis of cells was performed using a 488 nm argon laser EPICS XL-MCL flow cytometer controlled by EXPO 32-ADC software (Beckman Coulter, Fullerton, CA, USA). ROS values were analyzed based on fluorescence intensity.
GSIS
Static incubation of the INS-1 rat insulinoma cell line in Krebs–Ringer buffer (KRB) (118 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.18 mmol/L KH2PO4, 1.18 mmol/L MgSO4, 25 mmol/L NaHCO3, 10 mmol/L HEPES, and 0.1% BSA, pH 7.4) containing either non-stimulatory or stimulatory concentrations of glucose (5.6 mM or 16.7 mM, respectively) was performed for 1 hour. Insulin levels in KRB media were collected from the static incubations from INS-1 cells using 95.5 ethanol: hydrochloric acid solution was measured using an enzyme-linked immunosorbent assay (Rat Insulin ELISA kit; Mercodia, Uppsala, Sweden).
Real time RT-PCR
Total RNA was obtained from INS-1 cells using Trizol Reagent (Bio Science Technology, Korea). cDNA was synthesized using 1 μg total RNA with oligo-(dT) primers and Prime RT Premix (GENET BIO, Korea). Real-time RT-PCR was performed in the Light-Cycler (Roche, Germany) as previously described. The following primers were used: insulin, 50-ACC CAA GTC CCG TCG TGA AGT-30 (forward) and 50-CCA GTT GGT AGA GGG AGC AGA TG-30 (reverse); for PDX-1,50-GGC TTA ACC TAA ACG CCA CA-30 (forward) and 50-GGG ACC GTC CAA GTT TGT AA-30 (reverse); CD36, 50-GTG GCT AAA TGA GAC TGG GAC C-30 (forward) and 50-AGA CCA TCT CAA CCA GGC CC-30 (reverse); β-actin, 50-TAC TGC CCT GGC TCC TAG CA-30 (forward) and 50-TGG ACA GTG AGG CCA GGA TAG-30 (reverse).
Palmitate uptake
Palmitate uptake was determined using a modified method reported by Wallin et al. (4). Briefly, cells were trypsinized and washed in ice-cold KRB in the absence of glucose. An equal amount of cells was transferred to new tubes and centrifuged. Cells were re-suspended in ice-cold KRB (11.1 mmol/L glucose) and 2.1 lCi 14C-palmitate (Perkin Elemer Life Science Inc., Boston, MA) and transferred to microcentrifuge tubes (50 lL), which were prepared with a bottom layer of 6 mol/L urea solution (20 lL) over layered by a 10:3 mixture of dibutyl-dinonylphthalate (200 lL). Uptake was terminated by centrifugation at 8,000 rpm for 15 minutes. The urea layer containing the cells was transferred into scintillation vials and radioactivity was measured after addition of scintillation cocktail (Aqueous Counting Scintillation, Amersham, Canada) using a liquid scintillation counter (Packard Bioscience Company, Meriden, CT, USA).
RESULTS
The mRNA expression of insulin was decreased in the high dose palmitate (1 mM) group, compared with the control group, whereas mRNA expression of CD36 and intracellular peroxide level were increased in INS-1 cells. However, no significant change was observed in glucose stimulated insulin secretion (GSIS) by palmitate (data not shown).
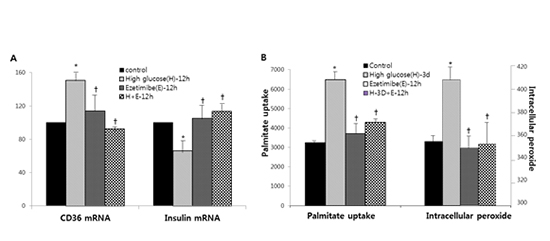
The effects of high glucose (30 mM) with or without ezetimibe for 12 hours on insulin secreting cells are shown in Fig. 1. The high glucose group showed increased CD36 mRNA expression and decreased insulin mRNA expression. Ezetimibe per se in normal glucose media induced no changes in mRNA expression of CD36 and insulin. However, treatment with ezetimibe in high glucose showed that the increased CD36 mRNA expression in high glucose was suppressed and decreased insulin mRNA expression was reversed with ezetimibe (Fig. 1A and 1B). Similar results were observed again in primary rat islet cells (Fig.1C and 1D).
Fig. 1
The effects of high glucose (30 mM) with or without ezetimibe on insulin secreting cells. The mRNA expression of insulin was decreased with high glucose (H-12h), which was reversed by ezetimibe (H+E-12h) in INS-1 cells (A) and rat islets (C). CD36 mRNA expression was increased with high glucose (H-12h), but decreased by ezetimibe (H+E) in INS-1 cells (B) and rat islets (D). Bars are mean ± SE of three separate experiments. *
P < 0.05 vs. Control, †
P < 0.05 vs. H-12h treated cells. 12h, 12 hours.

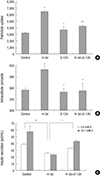
Three-day exposure of INS-1 cells to 30 mM glucose for induction of glucotoxicity resulted in an increase in palmitate uptake which was decreased by treatment with ezetimibe (Fig. 2A). An increase in Intracellular peroxide level and a decrease in GSIS were induced with three-day exposure of high glucose; however, ezetimibe induced a significant decrease in intracellular peroxide level and reversal of GSIS (Fig. 2B and 2C).
Fig. 2
The effects of three-day exposure of INS-1 cells to 30 mM glucose. Palmitate uptake (A) and intracellular peroxide levels (B) following an exposure to high glucose conditions (H-3d) for 3 days were significantly elevated, which were decreased by the ezetimibe (H-3d+E-12h) in INS-1 cells. Decreased glucose stimulated insulin secretion (GSIS) (C) by high glucose (H-3d) was reversed by ezetimibe (H-3d+E-12h). Bars are mean ± SE of three separate experiments. *P < 0.05 vs. Control, †
P < 0.05 vs. H-3d. 3d, 3 days; 12h, 12 hours.
DISCUSSION
Findings of the present study demonstrated that ezetimibe reversed high glucose induced increased CD35 expression, palmitate influx, and ROS levels and also increased insulin secretion in INS-1 cells and primary rat islet cells. There is a controversy regarding whether or not the effect of elevated FAs on pancreatic beta cells is beneficial (16-19). However, it is widely accepted that prolonged exposure to elevated FAs along with high glucose causes pancreatic β-cell dysfunction and apoptosis, resulting in glucolipotoxicity (1). Findings of the present study demonstrated that a high level of FAs led to an increase in oxidative stress and a decrease in insulin mRNA expression, but did not cause impairment of GSIS. The reasons why the effects of FAs on beta cell dysfunction in vitro have been various are the differences of sensitivity to the cytotoxic effects of FAs between clonal cells and primary beta cells, the disparity of concentration of FAs used in vitro, and lack of concern for the concentration of potent FAs fraction unbound to bovine serum albumin (BSA) (1).
The finding that high glucose condition in normal FA concentration increased intracellular uptake of FAs was shown again in the present study along with our previous study (5). This finding implies that the conditions inducing high FA influx may result in insulin secretory dysfunction in pancreatic beta cells. In addition, the finding that high dose FAs or glucose both induced an increase of ROS demonstrates the possible involvement of oxidative stress in FAs-induced beta cell dysfunction. This is consistent with the report (20) that high FAs or glucose-induced beta cell apoptosis was prevented by antioxidants, so that these cytotoxic effects could be mediated by oxidative stress. Mechanisms other than oxidative stress have been proposed to mediate fatty acid-induced beta cell dysfunction, including altered intracellular lipid partitioning, ceramide formation, activation of protein kinase C, inflammatory mechanisms, and endoplasmic reticulum (ER) stress; and glucotoxicity and lipotoxicity are believed to induce a synergistic loss of beta cells (1,21-25).
CD36 plays an important role in induction of fatty acid influx in pancreatic beta cells. One study demonstrated that induction of CD36 over-expression in INS-1 cells resulted in an increase in fatty acid uptake and attenuation of the normal potentiating effect of fatty acids on glucose stimulated insulin secretion (4). Findings of the present study demonstrated that even in normal fatty acid condition, high glucose induced expression of CD36 with increasing fatty acid influx and ROS was accompanied by a decrease in insulin secretion. Therefore, it is proposed that regardless of concentration of fatty acids, insulin secreting cells exposed to high glucose show increased fatty acid uptake resulting in beta cell dysfunction, at this time, CD36 plays an important role. It is suggested that elevated levels of glucose or fatty acids might regulate partitioning of calories into cells by induction of genes like CD36 that promote fatty acid uptake and triglyceride deposition, which contribute to acceleration of insulin resistance, cellular dysfunction, and atherosclerosis (26,27). Several studies have demonstrated that suppression of CD36 expression in insulin producing cells resulted in decreased fatty acid uptake and increased insulin secretion (28,29), and our previous study also demonstrated that suppression of CD36 by transfection with CD36 siRNA resulted in decreased oxidative stress and improved glucose stimulated insulin secretion in INS-1 cells (5). These findings suggested that inhibition of CD36 could be a target for prevention of glucotoxicity in pancreatic beta cells.
Ezetimibe blocks absorption of Niemann-Pick C1-Like 1 (NPC1L1)–mediated cholesterol in the apical brush border membrane of jejuna enterocytes (6). Recently, ezetimibe has also been reported to decrease the surface expression of CD36 (7-9). Therefore, ezetimibe seems to be able to prevent beta cell dysfunction by inhibition of CD36. The present study demonstrated that ezetimibe induced a decrease in expression of CD36, fatty acid uptake, and ROS, which were elevated in high glucose condition, and reversed the suppressed insulin secretion in INS-1 cells and primary rat islet cells. These findings are consistent with those of other studies reporting that ezetimibe-treated db/db mice showed improvement of the first phase of insulin secretion, reduced β cell loss, and protected function of β cells (13), and chronic administration of ezetimibe in OLETF rats was effective in glycemic control and lowering free fatty acids accompanied with preservation of pancreatic beta cell mass (14). According to one report, ezetimibe could inhibit the activity of hepatic NPC1Ll, reducing oxidative stress and ER stress, and then improve insulin resistance in the liver, resulting in glycemic control (12). However, few studies have reported on the mechanism underlying the effect of ezetimibe on pancreatic islets, saving the study reported by Yang et al. (14), in which ezetimibe induced a decrease in the activity of serum dipeptidyl peptidase-4 (DPP-4) and an increase in serum active glucagon, like peptide-1 (GLP-1) in OLETF rats, suggesting involvement of GLP-1 in the ezetimibe-mediated effects on pancreatic beta cells. However, there was an inconsistent report that treatment with ezetimibe in obese men did not affect the serum glucose-dependent insulinotropic polypeptide (GIP) and active GLP-1 (30). Therefore, results of the present study demonstrated that ezetimibe may have a beneficial effect on glucotoxicity in insulin secreting β-cells through inhibition of fatty acid influx via CD36.
In summary, high glucose increased CD36, fatty acid influx, and oxidative stress, resulting in impairment of insulin secretion, and ezetimibe reversed the insulin secretory dysfunction through inhibition of fatty acid influx via CD36 in pancreatic beta cells. Therefore, elevation of CD36 resulting from hyperglycemia, even at normal fatty acid level, may induce beta cell dysfunction through increase of fatty acid influx; it is proposed that aggressive correction of hyperglycemia with dyslipidemia may be needed in order to prevent glucotoxicity, even in diabetic patients with normal blood lipid levels.
 XML Download
XML Download