

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Bartter's syndrome and Gitelman's syndrome (GS) are inherited renal tubular disorders leading to increased urinary loss of sodium and potassium, low blood pressure, and metabolic alkalosis. Bartter's syndrome is a disorder of the thick ascending limb (TAL), caused by mutations in genes encoding sodium, potassium, or chloride transporters normally expressed in TAL (123). Clinical manifestations such as polyhydramnios, failure to thrive, polyuria, and salt craving begin to appear as early as in neonatal period. Among its subtypes, classical Bartter's syndrome (cBS) is caused by mutations in a gene encoding basolateral chloride channel type B (CLCNKB) that is expressed in cortical TAL and distal convoluted tubule (DCT) (1). cBS frequently shows phenotypic overlap with GS. Some cBS patients may show clinical features of GS, such as hypomagnesemia and hypocalciuria.
GS is caused by loss-of-function mutations in SLC12A3 encoding thiazide-sensitive sodium-chloride cotransporter (NCC) of the initial DCT (4). In contrast to Bartter's syndrome, GS is known to be characterized by low urinary calcium excretion and more frequent hypomagnesemia. Although it was once regarded as a milder variant of Bartter's syndrome, GS is clearly associated with significant disabling symptoms and low quality of life (5). Most of the mutations in SLC12A3 are missense and nonsense mutations, but frameshift, splice-site, and deep intronic mutations have been described as well (67). Although GS is an autosomal recessive disorder, homozygous mutations are found in only 18% of patients (8). More than 45% of GS cases have compound heterozygous mutations, 30% have single heterozygous mutations, and 7% have three or more mutations (9).
Although many mutations leading to the loss of function have been reported for each sodium transporter, the impact of gene diagnosis on clinical management of adult patients with GS and/or cBS is not always straightforward for several reasons. First, there is a significant overlap between GS and cBS in clinical manifestations and laboratory findings. Mutations in CLCNKB, in particular, seem to be responsible for mixed Bartter-Gitelman phenotype or at least be involved in a switch in clinical phenotype (1011). Second, gene diagnosis is not always immediately available in clinics. Nevertheless, even without a genetic report, clinicians should be able to make correct clinical diagnosis, predict long-term prognosis, and manage the condition accordingly. Third, the mechanism of disruption of transporter activity is not well understood, and it is not always possible to correlate a genotype with severity of disease. Finally, therapeutic measures based on specific mutation profile (i.e. gene therapy) are not available yet, diminishing the significance of mutation detection in terms of practical treatment. In this regard, it is always important to observe each patient carefully at a clinic and describe the natural history and prognosis when a clinician manages the patient with GS or cBS.
To investigate long-term prognosis and genotype-phenotype correlation, we followed patients who were referred for the diagnosis and management of unexplained chronic hypokalemia and metabolic alkalosis. Clinical diagnosis was made by history and laboratory findings. DNA sequencing was done to confirm clinical diagnosis. Long-term follow-up and management was done at the same clinic.
MATERIALS AND METHODS
Study participants and evaluations
At our outpatient nephrology clinic, we examined patients who visited for further evaluation of chronic unexplained hypokalemia and metabolic alkalosis. Careful history taking and physical examination were performed to exclude any conditions mimicking GS such as surreptitious diuretics use, cyclic vomiting, laxative abuse, chronic chloride deficient diet. Blood pressures were determined by office sphygmomanometer. We measured serum electrolytes, urea, and creatinine, and 24-hour urine excretion rates of sodium, potassium, chloride, calcium, magnesium, and creatinine. Hypocalciuria was defined as molar urinary calcium-to-creatinine ratio below 0.2 (calcium/creatinine [mg/mg] <0.07) from 24-hr urine collection. Fraction of excreted magnesium (FEMg) was calculated using the following formula: FEMg=(urine Mg×plasma Cr×100)/(0.7×plasma Mg×urine Cr)
Mutations in SLC12A3 and CLCNKB were detected by Sanger sequencing cDNAs and confirmed by sequencing the corresponding genomic region. Exons of SLC12A3 were sequenced as reported previously (12). In brief, both RNA and genomic DNA were isolated from peripheral-blood leukocytes. RNA was reverse transcribed and most parts of the coding sequences of SLC12A3 were sequenced after nested PCR. Exons 1, 2, and 3 of SLC12A3 were not covered by this nested PCR and therefore were directly sequenced from genomic DNA. For CLCNKB analysis, we amplified exon sequences by using sixteen pairs of primers against the intron sequences flanking each exon. We used NM_000085.3/NP_000076.2 as reference sequences for CLCNKB/ClC-Kb (13) (Table 1).
After diagnosis, the patients were followed up at our nephrology clinic. Liberal salt intake was encouraged and oral potassium and magnesium were titrated to achieve normal plasma concentrations and restore normal acid-base status. A potassium-sparing diuretic was added to the regimen in case of intractable hypokalemia.
RESULTS
Patients' demographic and clinical characteristics
In this cohort, we enrolled 34 patients from 31 families (Table 2). Male-to-female ratio was approximately 2:1 (23:11), and the median age at the time of presentation was 24.5. Low-extremity weakness and/or paralysis were the most common symptoms. One patient was found to have hypokalemia when she suffered a cardiac arrest during general anesthesia. All patients had low-to-normal blood pressure (systolic blood pressure 111.2±10.9 mmHg, diastolic blood pressure 69.0±8.5 mmHg, mean±standard deviation), hypokalemia (2.9±0.3 mM), and metabolic alkalosis (arterial pH 7.45±0.05, plasma bicarbonate 30.2±3.7 mM). Urinary biochemistry revealed renal wasting of sodium, potassium, and chloride (24-hour urine sodium, potassium, and chloride: 190.1±76.7 mM/d, 103.2±77.1 mM/d, and 241.3 ±166.3 mM/d, respectively). Male patients had earlier onset of neuromuscular symptoms (age of onset, male 21 vs. female 33 yr, P=0.04), and female patients had more severe hypokalemia (3.0±0.3 mM in male vs. 2.7±0.4 mM in female, P=0.04). Otherwise, there was no significant difference in plasma and urine electrolyte concentrations between male and female patients.
Mutational analysis and correlation with biochemical profiles
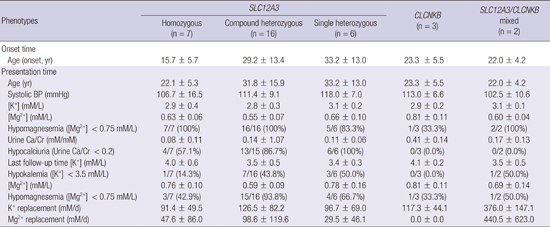
DNA sequencing identified 33 different mutations in SLC12A3 and 5 different mutations in CLCNKB (Table 3). For SLC12A3 mutations, 25 (75.7%) were missense, 4 (12.1%) splice-site mutations, and 4 (12.1%) insertion/deletion mutations causing frameshifts. We found 10 novel mutations in SLC12A3 (c.536T>A, c.784_785insGGCGTGGTCTCGG, c.964+1G>A, c.1174A>C, c.1762delG, c.1897_1898insG, c.2099T>C, c.2243C>T, c.2359C>T, and c.2369-4G>A). Of 31 patients with 1 or more mutant SLC12A3 alleles, 7 (22.5%) were homozygotes, 16 (51.6%) were compound heterozygotes, and 8 (25.8%) were single heterozygotes. Interestingly, c.2738G>A in SLC12A3 had been previously reported as overrepresented in hypertensive population (14). In our series, however, the patient with this genotype did not develop hypertension in the follow-up observation. Compared with patients with 1 mutant SLC12A3 allele, patients with 2 mutant SLC12A3 alleles had more severe hypomagnesemia (0.56±0.07 vs. 0.65±0.09 mM, P=0.03), but did not have more severe hypokalemia (2.8±0.4 vs. 3.1±0.2 mM, P=0.07). Five patients had 1 or more mutations in CLCNKB, of whom 3 (60%) had homozygous mutations and 2 had single heterozygous mutations. All of the CLCNKB mutations were missense, and 4 mutations were novel. Two patients had mutations in both SLC12A3 and CLCNKB.
Hypocalciuria and hypomagnesemia have been used as important clues in differential diagnosis in GS and cBS in pediatric patients (15). We assessed the usefulness of these parameters in adult patients. In our series, 27 (87.1%) out of 31 patients with SLC12A3 mutations had hypocalciuria (molar urinary calcium-to-creatinine ratio <0.2), whereas none of patients with mutations only in CLCNKB had hypocalciuria. Hypomagnesemia (plasma [Mg2+] <0.75 mM/L) was present in 30 (96.8%) of 31 patients with SLC12A3 mutations and in 1 (33.3%) of 3 patients with mutations only in CLCNKB. Hypomagnesemia was associated with varied degree of urinary Mg2+ loss, as calculated by FEMg. These findings were consistent with existing knowledge of GS and cBS, indicating that these biochemical criteria can also be useful in the diagnosis of GS in adult patients.
Response to the management
The patients were followed up at our nephrology clinic for 34.0±35.3 months. Follow-up management revealed wide inter-individual variability in daily amount of potassium and magnesium required to maintain plasma [K+] and [Mg2+] close to normal range (Table 4). Patients with 2 mutant SLC12A3 alleles did not have more severe phenotype than patients with 1 mutant SLC12A3 allele. There was no significant difference in plasma [K+], plasma [Mg2+], daily K+, or daily Mg2+ requirements (2 mutant SLC12A3 alleles vs. 1 mutant SLC12A3 allele: 3.7±0.6 vs. 3.4±0.3 mM, P=0.23 for plasma [K+]; 1.6±0.3 vs. 1.8±0.4 mM/L, P=0.08 for plasma [Mg2+]; median K+ doses, 96 vs. 120 mM/d, P=0.59; median Mg2+ doses, 58 vs. 19 mM/d, P=0.64). In contrast to the previous report (16), there was no difference between male and female in plasma [K+] (male vs. female, 3.6±0.5 vs. 3.7±0.7 mM, P=0.89), plasma [Mg2+] (0.65±0.18 vs. 0.66±0.20 mM, P=0.42), potassium requirements (median K+ doses, 134 mM/d vs. 113 mM/d, P=0.87), and magnesium requirements (median Mg2+ doses, 59 vs. 38 mM/d, P=0.90).
We identified 2 patients with concomitant SLC12A3 and CLCNKB mutations (patient 30 and 31 in Table 2). In these patients, we noted a difference in plasma and urine biochemistry and amounts of oral potassium and magnesium required to normalize their plasma [K+] and [Mg2+]. Patient 30 was a 19-year-old male with a splice-site heterozygous mutation in SLC12A3 and a missense heterozygous mutation in CLCNKB. He had a long history of low-extremity weakness and polyuria. At the time of presentation, he had hypokalemia (3.0 mM/L), severe hypomagnesemia (0.58 mM/L), and low urinary calcium excretion rate (urine calcium-to-creatinine ratio 0.07). In the clinical follow-up, high-dose oral potassium (480 mM/day) and magnesium (880 mM/day) supplement combined with amiloride (20 mg/day) did not normalize plasma [K+] and [Mg++]. In summary, clinical manifestations and severe biochemical abnormalities including intractable hypokalemia, hypomagnesemia, and hypocalciuria in this patient were more consistent with severe GS. Patient 31 was a 25-yr-old male who was found to have a heterozygous mutation in SLC12A3 and a homozygous nonsense mutation in CLCNKB. He had had failure to thrive, polyuria, and polydipsia since the age of 3 yr. At the time of presentation, he had polyuria, polydipsia, hypokalemia (3.1 mM), moderate hypomagnesemia (0.63 mM), and normocalciuria (urine Ca/Cr ratio 0.26). He maintained his [K+] within normal range with oral potassium (272 mM/day) and spironolactone (50 mg/day), and did not require oral magnesium. His clinical manifestations were more consistent with cBS rather than GS. Clinical features and therapeutic response according to genetic abnormalities were summarized in Table 5.
DISCUSSION
We collected clinical and genetic information from 34 adolescent and adult patients with chronic hypokalemia and metabolic alkalosis. Mutations in SLC12A3 and CLCNKB were discovered by Sanger sequencing of exonic regions of each gene. We followed these patients with long-term outpatient observation and management, noting that there is little correlation between mutation profile and clinical course, including the severity of electrolyte imbalance at presentation and after treatment, amount of potassium and/or magnesium replacement, response to treatment, and subjective symptoms.
Abnormalities in divalent cation metabolism were the most important clinical clues in our differential diagnosis of GS. Hypocalciuria and hypomagnesemia were strongly associated with SLC12A3 mutations. Although the mechanism underlying hypocalciuria and hypomagnesemia in GS remains unclear (17), urinary Ca2+ excretion rate and plasma [Mg2+], as shown in pediatric patients, can be useful markers for differential diagnosis in GS and cBS in adults (15). Urinary excretion of Mg2+, as calculated by FEMg, was not uniformly higher than 2%, which is a threshold generally accepted for a renal cause of Mg2+ loss in hypomagnesemia. This may reflect the fact that our patients were evaluated at a tertiary referral center, and that most patients were likely receiving treatment for hypomagnesemia at the time of the evaluation.
An unexpected finding of a homozygous mutation of c.2738G>A in a GS patient may point to the temporal change of phenotype in GS patients. Homozygotes of this genotype were originally reported to be associated with essential hypertension (14). This case may represent the unexpectedly high incidence of essential hypertension found in a recent retrospective study of GS patients (16). There may be a transition in clinical phenotype from low blood pressure to high blood pressure as the patient ages, with a tendency to develop hypertension overriding the defect in sodium-chloride cotransporter. Long-term follow-up is warranted to better understand the natural history of this genotype.
Gender or the number of mutated SLC12A3 alleles was not associated with significant difference in clinical phenotype. We need to interpret with caution some findings that were significantly different between male and female or between patients with different number of mutated alleles. Recall of symptoms was not likely to be perfect; Plasma [K+] and [Mg2+] and requirements for oral potassium and magnesium depend on many factors including intake, other causes of electrolyte loss, and the use of other medications.
Although genetic screening has been used as a gold standard for the diagnosis of inherited hypokalemic renal tubular disorders, it is not clear if it has a critical role in managing patients and predicting their prognosis. Our patient series showed that there is little difference in clinical course between patients with different mutations in SLC12A3 and CLCNKB, suggesting that there is little need to pursue sophisticated genetic diagnosis in practical care. Our finding is also consistent with the notion that Bartter- and Gitelman-like syndromes can be better understood in terms of the functional status of involved tubule segments than in terms of specific genes harboring mutations (18). In this classification scheme, GS and cBS can be collectively defined as the "dysfunction of the DCT" that can be mimicked by thiazide diuretics (18). In this regard, diuretic loading test has been successfully used in a nephrology ward to assess the functional status of sodium transporters, though it is associated with the risk of hyponatremia and dangerous hypokalemia and the result of the test is sometimes difficult to interpret (1920).
Two cases of concomitant mutations in SLC12A3 and CLCNKB further complicate our interpretation of genotype-phenotype relationship. The clinical picture of patient 31, a 25-yr-old male patient who had homozygous mutations in CLCNKB and a heterozygous mutation in SLC12A3, was more consistent with cBS. He had early-onset manifestations such as failure to thrive, polyuria, and polydipsia; and hypocalciuria was not as prominent as in typical GS patients. In this case, as previously reported in cases in kindred (21), homozygous mutations in CLCNKB seemed to determine the overall status of sodium and chloride reabsorption in the TAL and DCT, and the role of the nucleotide variant in SLC12A3 was unclear. However, further clinical follow-up is warranted because patients with CLCNKB show overlapping clinical manifestations and biochemical profiles with GS and some of them show transition in clinical phenotype from cBS to GS (1011). In contrast, Patient 30, a 19-yr-old male patient with a heterozygous mutation in SLC12A3 and a heterozygous mutation in CLCNKB, presented with hypocalciuria and severe intractable hypokalemia and hypomagnesemia, more suggestive of severe form of GS rather than cBS. It is possible that the differences of genotype, such as homozygous mutation vs. single heterozygous mutation in CLCNKB could determine the differences of phenotype into GS or cBS.
There are several limitations in our genetic study. We did not attempt to sequence other genes involved in the pathogenesis of Bartter's syndrome such as SLC12A1 or KCNJ1, because the clinical manifestations and laboratory findings were much more consistent with GS. We did not report deep intronic mutations or large deletions of one or more exons because of the limitation of exonic DNA-based sequencing. Deep sequencing of intronic regions or multiplex ligation-dependent probe amplification can be helpful in this regard, as large genomic rearrangements are suggested as a possible mechanism of GS in single heterozygotes (22). Finally, more detailed information from family trees could have helped us identify pathogenic mutations more correctly, but it is usually not possible to collect complete information about family history based on outpatient visits.
In summary, our collection of clinical and genetic data from 34 patients with chronic hypokalemic metabolic alkalosis shows wide variability in clinical phenotype. Hypocalciuria and hypomagnesemia are important clinical markers for differential diagnosis between GS and cBS. Variations in genotype are not correlated with clinical course or electrolyte requirements. More data from molecular biology and clinical long-term observation are needed to answer the question of phenotypic variability of SLC12A3 and CLCNKB mutations.





 XML Download
XML Download