

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Coagulation factor V (FV), known as labile factor or proaccelerin, plays a pivotal role in the blood coagulation cascade by having both procoagulant and anticoagulant functions (1). FV is cofactor for the prothrombinase complex that activates prothrombin to thrombin and interacts with several coagulation factors and also modulator in the anticoagulant pathway by downregulating factor VIII activity. FV is mainly synthesized by liver and present mostly in plasma, accounting for approximately 80% of the circulating FV (2). In addition to plasma FV, 20% of the total FV in whole blood is found within α-granules of platelets (12). Although possibly megakaryocytes can synthesize FV, the majority of platelet FV originate from plasma through a mechanism of plasma FV uptake (endocytosis) by bone marrow megakaryocytes (13). As consequence, the levels of FV are affected by liver synthetic function, and FV deficiency can lead to bleeding as a result of physiologic role of FV in contributing thrombin generation.
FV deficiency is a rare bleeding disorder (8.3% of all rare inherited bleeding disorders) with an estimated incidence of 1 in 1,000,000 (3-5), which is associated with a variable spectrum of bleeding manifestations ranging from mucosa and soft tissue bleeding (such as epistaxis and hemarthroses) to life-threatening hemorrhages. Until now, more than 200 cases of FV deficiency have been described in literature (35). Inherited FV deficiency, also known as parahemophilia or Owen disease, had an autosomal recessive trait, and occurred approximately up to 10 times more frequently in West Asian countries where consanguineous marriage are common (such as Iran and southern India) than in the Western world (4). In a North American registry for rare bleeding disorders published at 2004 (6), while the majority of patients with FV deficiency were Caucasian accounting for 67% of all patients, Asian had only 2% of affected patients. Because of the relative rarity of FV deficiency in East Asia, there have been only a few case reports in Korea, and data on genetic, laboratory, and clinical characteristic of this disorder have been limited. Herein, we analyzed the clinical-laboratory characteristics and treatment outcomes in 10 Korean patients with FV deficiency. To our knowledge, this retrospective study is the first research for Korean patients with FV deficiency.
MATERIALS AND METHODS
Study population and data collection
From January 1987, when FV deficiency was reported first in Korea, to December 2012, data was collected from PUBMED and Korea PUBMED database search using the term ‘factor V deficiency’, ‘factor V inhibitor’, and ‘Korea’. In addition, the case reports presented at Korea Society on Thrombosis and Hemostasis (KSTH) and the medical records in our institution were reviewed retrospectively. A total of 13 patients were identified from six case reports published in Korea (7-12) and seven case presentations in KSTH meetings including one case in our institution. Among them, three of seven case presentations were excluded due to incomplete data and 10 cases were finally enrolled in this study (Fig. 1). Data were collected on the age of the patient at the date of diagnosis, past medical history, family history, bleeding sites, number of bleeding events, laboratory findings, applied treatments, and outcomes. Laboratory findings included blood counts, plasma FV levels, prothrombin time (PT), activated partial thromboplastin time (aPTT), fibrinogen, bleeding time, mixing test, and FV inhibitor titer.
Definition of inherited and acquired FV deficiency
Typically, FV deficiency was suspected in a patient with bleeding symptoms who has a prolongation of both PT and aPTT. We excluded disseminated intravascular coagulopathy and liver disease. The patient with low FV levels who has a positive family bleeding history or no presence of FV inhibitor was defined as an inherited FV deficient-patient because FV deficiency is usually inherited as an autosomal recessive trait. Acquired FV deficiency was defined as the identification of acquired FV inhibitors resulting in usually a combined prolongation of PT and aPTT, which cannot be corrected after mixing with normal plasma (typically in a 1:1 ratio), and/or an isolated FV deficiency in a patient with an otherwise negative personal and family bleeding history.
Definition of severity and bleeding manifestations
By residual FV level, severe disease is defined as < 1% factor activity, whereas 1% to 5% and > 5% of normal are defined as moderate and mild disease. Variable bleeding symptoms such as mucous membrane bleeding, skin bleeding, intracranial bleeding, and peritoneal bleeding were classified as life-threatening and non-life threatening hemorrhage. Life-threatening bleeding episode was considered as any bleeding requiring blood transfusion or any intervention to control active hemorrhage. According to published criteria, recurrent epistaxis was defined as bleeding occurring spontaneously more than five times and last more than 10 minutes, and menorrhagia qualified when menstrual periods last at least 6 days and/or required the therapeutic use of hormonal preparations.
RESULTS
Patient characteristics
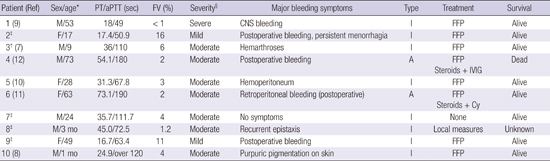
The clinical-laboratory characteristics of the 10 patients with FV deficiency are listed in Table 1. The median age of patients at the time of diagnosis, six males and four females, was 26 years (range, 1 month-73 years). In laboratory findings, the median time of PT and aPTT for these patients were 31.3 seconds (range, 16.7–73.1 seconds) and 110 seconds (range, 49–190 seconds), respectively, and the median plasma levels of FV at diagnosis was 3.5% (range, 0.9%–16%). Six of 10 patients (60%) were classified as moderate FV deficiency and three (30%) as mild FV deficiency. Only one patient (10%) had severe disease, showing FV levels 0.9%. Levels of plasma factor VIII were available for 60% of all cases. Among these cases, one had concurrent factor VIII deficiency, with 0.6% of factor VIII activity. Of our case, eight patients were diagnosed as inherited FV deficiency and familial screening test for factor deficiencies was performed in 6 of 8 cases, with no reported coagulation abnormalities in family members of patients. No fully correction on mixing test was found in two cases, one of which documented an acquired FV inhibitor titer of 3.5 Bethesda units (BU) determined with a Bethesda assay. One of these patients diagnosed with acquired FV deficiency had history of surgery for pancreatic head carcinoma and use of certain antibiotics (beta-lactam).
Table 1
Summary of the clinical and laboratory characteristics of the 10 Korean patients with FV deficiency

| Patient (Ref) | Sex/age* | PT/aPTT (sec) | FV (%) | Severity§ | Major bleeding symptoms | Type | Treatment | Survival |
|---|---|---|---|---|---|---|---|---|
| 1 (9) | M/53 | 18/49 | <1 | Severe | CNS bleeding | I | FFP | Alive |
| 2‡ | F/17 | 17.4/50.9 | 16 | Mild | Postoperative bleeding, | I | FFP | Alive |
| persistent menorrhagia | ||||||||
| 3† (7) | M/9 | 36/110 | 6 | Moderate | Hemarthroses | I | FFP | Alive |
| 4 (12) | M/73 | 54.1/180 | 2 | Moderate | Postoperative bleeding | A | FFP | Dead |
| Steroids + IVIG | ||||||||
| 5 (10) | F/28 | 31.3/67.8 | 3 | Moderate | Hemoperitoneum | I | FFP | Alive |
| 6 (11) | F/63 | 73.1/190 | 2 | Moderate |
Retroperitoneal bleeding (postoperative) |
A | FFP | Alive |
| Steroids + Cy | ||||||||
| 7‡ | M/24 | 35.7/111.7 | 4 | Moderate | No symptoms | I | None | Alive |
| 8‡ | M/3 mon | 45.0/72.5 | 1.2 | Moderate | Recurrent epistaxis | I | Local measures | Unknown |
| 9‡ | F/49 | 16.7/63.4 | 11 | Mild | Postoperative bleeding | I | FFP | Alive |
| 10 (8) | M/1 mon | 24.9/over 120 | 4 | Moderate | Purpuric pigmentation on skin | I | FFP | Alive |
Ref, reference; PT, prothrombin time; aPTT, activated partial thromboplastin time; FV, factor V; A, acquired; I, inherited; CNS, central nervous system; FFP, fresh frozen plasma; IVIG, intravenous immunoglobulin; Cy, cyclophosphamide.
*Age, age at diagnosis.
†First case report of FV deficiency in Korea.
‡Based on medical records of each case presented at meetings of Korea Society on Thrombosis and Hemostasis.
§Severe disease is defined as < 1% factor activity, whereas 1% to 5% and > 5% of normal are defined as moderate and mild disease, respectively.
![]()
Clinical manifestations and severity of FV deficiency
Nine (90%) patients had variable bleeding symptoms and only one patient was asymptomatic. Of the symptomatic patients, five (55.5%) were male and four (44.5%) were female. The major bleeding sites and severity of FV deficiency were shown in Table 2. The most frequent symptoms associated with FV deficiency were mucosal tract bleedings (40%) such as epistaxis, oral cavity hemorrhages, and menorrhagia in females. Menorrhagia occurred in two of three women of child-bearing age, who had severe or mild factor deficiency. Hemarthroses without clinical significance occurred in one patient with mild factor activity, and postoperative bleeding was found in four (40%) patients including three patients with moderate FV deficiency and one with mild FV deficiency. Post-operative bleeding was the first symptom of disease in three patients who had negative history of spontaneous bleeding. One patient who was detected incidentally on coagulation abnormalities had moderate FV plasma levels without any bleeding symptoms. Two patients presented within the first year of life. Four patients (40%) displayed life-threatening bleeding occurring in the peritoneal cavity (n = 2), central nerve system (n = 1), and retroperitoneal space (n = 1), respectively. Among them, intracranial bleeding occurred in a patient with severe FV deficiency, who was hospitalized for sudden onset of drowsy mental status resulting from hemorrhage in left thalamus and intraventricular hemorrhage in occipital lobe. Fatal retroperitoneal bleeding occurred in a moderate FV-deficient patient who had also a hematoma in left psoas muscle. No lethal hemorrhages happened to the patients with > 5% factor activity. Recurrent bleeding episodes were observed in four patients. Among these patients, three had frequent mucosal bleedings such as epistaxis and menorrhagia in females, and one had recurrent hemoperitoneum during ovulation, with FV activity being < 5%. Other symptoms such as gastrointestinal tract and hematuria were not identified. Two cases of moderate disease presenting with neonatal and perinatal bleeding were reported. Of these patients, one had a massive scalp hematoma and purpuric pigmentations in whole skin at birth and was finally diagnosed with FV deficiency combined with factor VIII deficiency. Vaginal delivery with median episiotomy was performed in a patient with severe inherited FV deficiency. During delivery, 6 unit of fresh frozen plasma (FFP) was transfused and after additional administration of FFP (7.5 mL/kg every 12 hours for two days), the patient was discharged on the fourth postpartum day without hemorrhage or hematoma.
Table 2
The bleeding sites and frequency of bleeding episodes in symptomatic patients

![]()
Treatment of patients with inherited FV deficiency
FFP was the primary treatment option in the majority of symptomatic patients with FV deficiency regardless of FV plasma levels. Asymptomatic patients required no treatment. In most instances, especially in severe bleeding, FFP was used before a diagnosis was confirmed, with initial dose of 15 to 20 mL/kg of body weight, adjusting the dosage on the basis of clinical symptoms and coagulation profiles. In addition to FFP replacement, local measures and/or administration of antifibrinolytic drug (tranexamic acid, orally or intravenously) were used for less severe mucosal bleeding and before invasive procedures episodically. Two female patients with persistent menorrhagia also received hormonal therapy with estrogen-progesterone preparations to reduce menstrual blood loss, resulting in little response. Prophylactic FFP infusions before invasive procedures such as tooth extraction (n = 2), during vaginal delivery (n = 1), and preoperatively (n = 1) were performed in affected patients, who developed no hematoma or postpartum hemorrhage thereafter. No treatment-related complications such as allergic reactions to plasma products, anaphylaxis, and fluid overload after repeated FFP infusions were founded in all reported cases. Overall, most FV-deficient patients in this study had a good treatment response for bleeding control and a favorable prognosis.
Treatment of patients with acquired FV deficiency
FFP was also first-line treatment in two acquired FV-deficient patients. Due to severe bleeding not controlled with FFP replacement, the follow-up therapies including the immunosuppression with corticosteroids plus cyclophosphamide and infusion of intravenous immunoglobulin (IVIG) were performed as salvage treatment. Also platelet concentrates were given repeatedly in these patients with no benefit for the bleeding control and correction of coagulation profiles. Among these patients, one patient with fatal retroperitoneal bleeding due to idiopathic acquired FV inhibitor showed significant improvement of FV activity and PT levels after taking cyclophosphamide (2 mg/kg/day) and methylprednisolone (1 mg/kg/day). However, the other patient who had confirmed FV inhibitors occurred after surgery for pancreatic cancer died of bleeding complications with increased quantified titer of inhibitor (8.4 BU) in spite of the treatment with corticosteroids plus IVIG (400 mg/kg/day for 5 days).
DISCUSSION
This is the first report to date of comprehensive analysis of FV deficiency in Korea, comprising eight inherited and two acquired FV-deficient patients. Whereas previous reports mainly focused on description of case series since 1987 when the first case was reported in Korea (7), in the present study, we demonstrate the clinical manifestations, laboratory findings, and treatment outcomes in 10 Korean patients with FV deficiency. Inherited FV deficiency occurs with an estimated prevalence of one per million individuals worldwide (3). Although no precise epidemiologic data existed for FV deficiency in Korea, only 10 patients were identified as having inherited or acquired FV deficiency during the study periods. However, because of lack of understanding about FV deficiency and ignorance of many minor bleeding episodes commonly by physicians, it is possible that the true incidence rate of FV deficiency in Korea is higher than expected. Due to rarity of this disorder, national comprehensive registry such as The North American registry for rare bleeding disorder in the USA (6) or The Hemophilia Centre Directors Organization in the UK (13) will be needed to gather data about disease prevalence, genotype information, clinical manifestations, treatment strategies and complications associated with disease or treatment.
In the literature, large series of FV deficiency usually involved both children and adults (35614). In the Iranian cohort (14), age distribution range was reported as 3 to 65 years and most patients had bleeding symptoms before the age of 6 years. The ages of our patients at diagnosis were ranging between 1 month and 73 years (median, 26 years). Two patients (20%) were diagnosed when they were younger than one year of age. Of the inherited FV-deficient patients in this study, only three had a positive family history for bleeding disorder, despite the autosomal recessive inheritance pattern for inherited FV deficiency.
FV deficiency may be presented with various bleeding patterns in affected patients; epistaxis, hemarthrosis, menorrhagia, gastrointestinal bleeding, hematuria, bleeding from umbilicus stump, and intracranial bleeding have been reported. Lak et al. (14) in a series of 35 Iranian patients reported that epistaxis and oral cavity bleeding were the predominant symptoms (57%). In this study, most patients had variable bleeding events and predominantly involved mucous membranes of oropharyngeal space and genital area in females, which is generally similar to the findings in the Iranian study. In contrast, other symptoms of mucosal bleeding such as hematuria and melena were not reported. Of our cases, one was asymptomatic and diagnosis was made by routine laboratory work-up. In the Iranian cohort (14), excessive post-operative bleeding (most often after circumcision) and CNS bleeding were presented in 43% and 6%, respectively. Additionally, Acharya et al. (6) had reported the frequency of CNS bleeding as 8%. In our study, four patients presented with fatal bleeding symptoms and among them, one experienced overt intracranial hemorrhage. Another hemorrhage type in our study was postoperative bleeding with a frequency of 60%. Accordingly, this pattern of bleeding symptoms in our study is generally similar to that described by other studies.
The severity of bleeding symptoms of FV deficiency is variable and generally reported that the correlation between plasma FV levels and clinical manifestation is not clear-cut (12). Many patients with undetectable levels of FV (< 1%) displayed only mild-to-moderate bleeding phenotype. Some patients with FV levels greater than 5% have severe bleeding symptom, even cases of the thrombosis have been reported (2). Among the moderate to severe FV-deficient patients (n = 7) in our study, whereas four displayed fatal bleeding episodes, two had mild bleeding symptoms and one had no bleeding symptom. These observations of diverse clinical phenotype associated with FV levels strongly suggest the existence of additional factors modulating the bleeding diathesis in FV deficiency. As a possible explanation for these discrepancies, the recent studies identified the phenotypic modifiers of the bleeding predisposition in the severe FV-deficient patients, including platelet factor V, plasma tissue factor pathway inhibitor (TFPI), and pseudo-homozygous activated protein C resistance (2). Currently, several studies show that residual platelet FV may be crucial in maintaining adequate hemostasis for patients with undetectable plasma FV levels (12), where it might explain the discrepancy between FV levels and bleeding manifestations. In our study, platelet FV was not routinely evaluated, especially in severe FV-deficient patients, reporting only plasma FV levels, and we did not perform genotype and molecular analysis of FV molecules because of lack of specialized research laboratories to identify the molecular and/or genetic defects in patients with FV deficiency.
Because no FV concentrates are currently available, the mainstay of FV deficiency treatment in the presented study was infusion of FFP, which is widely available and relatively inexpensive (23). FFP was used in 80% of patients, and, in most instances, they were given it before a diagnosis was confirmed. In our study, most congenital FV-deficient patients had a good treatment response for bleeding control and a favorable prognosis after single or repeated FFP administration, without any complications such as volume overload. In addition to replacement therapy, local measures or antifibrinolytic agents such as tranexamic acid alone or in combination with FFP administration may be useful in the management of the less severe bleeding cases (4). Also combined oral contraceptives (COC) were used in two female patients with persistent menorrhagia to reduce menstrual blood loss and little response for bleeding control was observed.
Acquired FV deficiency associated with inhibitors is a rare event and may cause significant morbidity and mortality. Recently, about 150 cases of acquired FV deficiency, with only two Korean cases, have been reported in literatures (15). In Korea, the first case was described in 2008 (12). Variable risk factors previously reported to be associated with the occurrence of FV inhibitors included surgical procedures, antibiotics administration such as the beta lactams, blood transfusions, malignancy, autoimmune diseases, and exposure to bovine thrombin (16). In our study, two patients had acquired FV deficiency with either idiopathic etiology or associated risk factors for the presence of FV inhibitors. Among them, one patient with acquired FV deficiency had proven inhibitors after cancer surgery, which may play a causative role in the development of the inhibitor. Also, the perioperative use of antibiotic (second-generation cephalosporin) and/or old age may be contributed to develop the FV inhibitors, being difficult to establish. Another patient with no definite risk factors, mentioned above, is described as idiopathic. The clinical phenotype of patients with acquired FV deficiency might vary from asymptomatic laboratory abnormalities to fatal bleeding (1516). Our study showed that life-threatening bleedings including postoperative hemoperitoneum and retroperitoneal hemorrhage were observed in all patients with acquired FV inhibitors with one fatality among them, and both of them had severe FV deficiency with plasma FV levels of 2%. Ang et al. (16) showed that in a retrospective review of 76 acquired FV-deficient patients with proven FV inhibitors, low FV levels (median 1%, range of 0%–23%, P = 0.068) was associated with a tendency for the severe bleeding compared with non-bleeders, with no statistical significance. In our study, all acquired FV-deficient patients had plasma FV levels of 2%, and showed lethal bleeding events, which findings were consistent with previous studies.
Generally, the treatment strategy of acquired FV inhibitors included the bleeding control and the eradication of the FV inhibitors, especially in symptomatic patients (1516). Variable hemostatic agents were used in attempts to control clinically severe bleeding events. They included FFP, platelets, prothrombin complex concentrate (PCC), recombinant activated factor VII (rVIIIa), with response rate of 15%, 69%, 80%, and 33%, respectively (15). Especially, a consistent number of patients in published literatures had a significant clinical response (69%–71%) to transfusion of platelet concentrates which appear to be resistant to inhibitors until platelets are activated (15). However, our patients with the inhibitors showed no response to repeated platelet transfusion and received no additional hemostatic agents such as PCC or rVIIa. In addition to the control of the bleeding, to eradicate the inhibitors and suppress production of the autoantibody, a number of therapeutic options including immunosuppressant, immunoadsorption, IVIG, and plasmapheresis have been used in bleeding patients with acquired FV inhibitor with varying success (16). In a systemic review by Ang et al. (16), 15 patients with FV autoantibodies were treated with steroids plus chemotherapy, including vincristine, cyclophosphamide, doxorubicin, and chlorambucil, with a response rate of 86.6%. Among two patients with FV inhibitor in our study, one of them developed massive retroperitoneal bleeding, and initially received FFP and platelet transfusion without response. Subsequently, immunosuppressive chemotherapy with high-dose steroids and cyclophosphamide was performed with the resolution of the FV activity on 4 weeks from start of treatment. However, the other patient who received corticosteroids and IVIG had no response with increased autoantibody titers and finally died of bleeding complications.
This study has potential limitations. Due to extreme rarity of the disease, limitations of this study were stemmed mostly from the small number of patients included and its retrospective nature, making it difficult for any solid conclusions to be drawn from these results. In addition, we could not analyse the genotype and molecule of FV in routine practice due to lack of specialized research laboratories to identify the molecular and/or genetic defects in FV-deficient patients. Despite of these inherent limitations, this study provides clinical data of Korean FV deficient-patients because FV deficiency is extremely rare disease and large clinical studies regarding this disease have not been reported in Korea.
In conclusion, despite its retrospective design and the small sample size, we found that Korean patients with FV deficiency had similar clinical manifestations and treatment outcomes shown in several previous studies. Mucosal tract bleedings were the predominant symptoms in FV deficient-patients and severity of bleeding symptoms were not always correlated with plasma FV levels. Most patients responded well in FFP replacement treatment, except the patients having an inhibitor to FV, who received an immunosuppressive therapy. Further studies with larger numbers of patients are warranted to define the actual incidence, clinical features, and appropriate therapeutic strategies and to clarify the accuracy of our results.
 XML Download
XML Download