

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Opioids are currently the most effective treatment option for cancer pain. Opioid analgesics for the treatment of pain allow normal living in 85% to 95% of patients (12). Considering their safety and efficacy, opioids should be administered daily to patients with moderate to severe cancer pain (3). Although morphine is the most commonly used agent for moderate and severe cancer pain (4), the use of alternative opioids is increasing due to adequate and safe pain relief, lower side effects, as well as convenience and low toxicity in long-term use (5).
Hydromorphone, a semi-synthetic μ-opioid agonist, is one such alternative opioid. It has been used extensively in the treatment of post-surgical pain (678), and has been proven a good alternative to morphine due to its excellent efficacy and tolerability in severe cancer pain (910). In order to provide sustained pain relief, conventional immediate-release tablets should be administered every 4 hours (11). However, such a high frequency of administration is inconvenient for patients and reduces their quality of life (12). Therefore, a sustained-release formulation of hydromorphone using push-pull active osmotic technology was developed for treatment of chronic pain with a single daily dose. Push-pull active osmotic technology was developed by ALZA Corporation (Mountain View, CA, USA). The system was designed to release a drug over a period of 24 hours, providing long-lasting analgesia (131415). The dosage form actively controls the release of the drug, and surrounding factors like pH or gastric motility do not significantly influence drug release (161718). Once-daily osmotic extended-release oral delivery system (OROS®) hydromorphone has the advantages of convenience, effectiveness, safety, decreased use of rescue medication (which may suggest a reduction in overall breakthrough pain), and quality of life benefits (14192021).
To date, approximately 1,500 subjects have participated in clinical trials on OROS hydromorphone (2022). Of these subjects, about 300 have been involved in clinical trials on cancer patients. Although the ingredient hydrochloric hydromorphone was developed in 1926, there is little data on the clinical usefulness of hydromorphone for chronic cancer pain control (21232425). The objective of this observational study was to evaluate the effectiveness of OROS hydromorphone in the management of cancer pain by measuring changes in pain intensity, levels of sleep disturbance, breakthrough pain, and end-of-dose failure before and after treatment as well as investigate patient satisfaction with this drug and overall assessment of drug effectiveness by the investigators.
MATERIALS AND METHODS
Patients
In this study, patients with cancer pain of moderate to severe intensity received OROS hydromorphone for 4 weeks. Subjects had to be capable of complying with the overall study course based on the judgment of the investigators. Exclusion criteria included: 1) use of OROS hydromorphone prior to participation in the study; 2) past or current abuse of drugs or alcohol; 3) pregnancy or women planning to become pregnant during the study period; 4) male subjects whose female partners did not use an effective contraceptive method (implant, injectable products, oral contraceptives, or intrauterine devices, etc.), and the male subjects were not infertile or not willing to practice abstinence during the study; 5) severe gastrointestinal diseases (diseases that could influence the absorption and metabolism of oral medication, such as dysphagia, vomiting, ileus, or severe enterostenosis); 6) hypersensitivity to hydromorphone; 7) use of monoamine oxidase inhibitors currently or within 2 weeks prior to administration of the study drug; 8) presence of conditions preventing subjects from participating in the study based on the warnings, precautions, and contraindications described in the package insert of the study drug and the judgment of the investigators.
Study design
This study was a multi-center, open-label, prospective, observational study conducted at 30 centers in the Republic of Korea. The duration of the study was 4 weeks. Demographic data were collected at the initial visit. Effectiveness and safety data were evaluated at visit 1 (baseline evaluation) and visit 2 (week 4; day 29 ± 7 days). Since this study was an observational study of routine patient care, the dose of OROS hydromorphone was adjusted by the investigators according to pain intensity and conditions of individual patients. Patients were started on a conservative dose and then the dose was adjusted according to adverse events and pain control. In patients currently receiving narcotic analgesics, the starting dose of OROS hydromorphone was based on the daily dose of previous narcotic analgesics using the percentage of the standard equivalent pain. For narcotics other than morphine, the total daily dose of OROS hydromorphone was calculated by using a conversion table after converting to morphine first. OROS hydromorphone tablets were administered by swallowing the whole tablet with a cup of water at almost the same time every day. Patients were instructed not to chew, break, or grind the tablet.
Assessment of effectiveness and safety
Pain intensity over a period of 24 hours was measured using a numerical rating scale (NRS; 0, no pain; 10, pain as bad as you can imagine). The degree of decrease in pain intensity score was the primary endpoint and calculated using the following equation:
Pain intensity difference (% PID) = (NRS at the baseline − NRS at final visit) / NRS at baseline × 100
Pain improvement was also assessed as secondary endpoints by measuring the change in pain intensity score over 24 hours and classifying the percentage decrease in pain intensity score by the type of previous analgesics used (non-opioid, weak opioid, or strong opioid). The level of sleep disturbance was assessed at visits 1 and 2 by measuring the mean frequency of waking during sleep due to pain over the past week. Breakthrough pain was assessed by measuring experience of pain, mean frequency of experiencing pain, and NRS scores for interference with performing daily activities due to breakthrough pain over the past week. End-of-dose failure over the past week was assessed based on the degree of interference with pain control. In addition, patient satisfaction with the study drug and detailed reasons for this were assessed at visit 2, and investigators’ overall assessment of drug effectiveness was recorded. Data of those NRS scores as well as patient’s satisfaction with drug and investigators global assessment of efficacy were totally obtained by an in-person interview at visits using questionnaires performed by investigators or clinical research coordinators in each centers. Details about questionnaires in Korean language were shown in supplementary file.
Safety analysis included all adverse events occurring during the period of treatment. Information on adverse events including onset date, seriousness, severity, outcomes, actions taken with regards to the study drug, and the relationship of the adverse event to the study drug as assessed by the investigators was described and recorded in the source document and case report forms of the subjects. Information on adverse events was collected via voluntary reporting by subjects or interview with subjects during visits. All adverse events were followed up until they were resolved satisfactorily or reached a clinically stable state.
Statistical analysis
Patient data were divided into 2 groups for analysis: a full analysis set (FAS) group and a safety group. Demographic data and effectiveness results were analyzed in the FAS group, and safety results were analyzed in the safety group. The safety group included patients who received at least one dose of the study drug. The FAS group was comprised of patients who received the study drug and underwent efficacy assessment at least once including visit 1 and excluded patients violating any of the inclusion/exclusion criteria. In the event of missing efficacy data for the visit at week 4, data for early termination were evaluated.
For the efficacy endpoints, descriptive statistics were presented for the values obtained at visits 1 and 2. The number of subjects and percentage frequency were presented for the frequency of waking during sleep due to pain, changes in the frequency of breakthrough pain for the past week, satisfaction of patients with the study drug, and overall assessment of drug effectiveness by the investigators. The parameters of primary and secondary endpoints were compared using Wilcoxon signed rank test. All data were analyzed with statistical analysis system (SAS, version 9.2, SAS Institute Inc., Cary, NC, USA) and a value of P < 0.05 was accepted as significant.
Ethics statement
All patients provided written informed consent before enrollment. The study was performed in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. The study was approved by the institutional review board of each medical institution and registered with ClinicalTrials.gov (Identifier: NCT 01273454).
RESULTS
Patients
A total of 648 subjects were enrolled from June 2009 to December 2009 (Fig. 1). Five hundred and seventeen subjects (79.8%) completed the study and 131 subjects (20.2%) were withdrawn from the study. Reasons for withdrawal included ‘loss to follow-up’ in 32 subjects (4.9%), ‘further study treatment unnecessary because pain was decreased or resolved’ in 31 subjects (4.8%), ‘lack of efficacy’ in 22 subjects (3.4%), ‘impossible to treat further due to occurrence of adverse events’ in 20 subjects (3.1%), and ‘other reasons’ in 26 subjects (4.1%).
 | Fig. 1Disposition of subjected patients.
A total of 648 subjects were enrolled from June 2009 to December 2009.
|
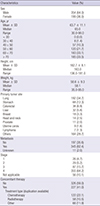
The characteristics of the 553 patients in the FAS group are summarized in Table 1. The median age of participants was 65.0 years and 64.0% of the patients were male. The most common primary tumor site was the lung (34.7%). Two hundred and twenty seven (41.0%) patients were receiving concomitant therapies for cancer such as chemotherapy (22.1%) and radiotherapy (10.5%). The initial dose administered was 11.5 ± 11.9 mg (mean ± standard deviation [SD]) and the dose at the completion of the study was 14.5 ± 18.7 mg. The total dose administered was 334.4 ± 408.5 mg and the mean duration of administration was 25.3 ± 9.1 days. The mean daily dose was 13.2 ± 15.7 mg. During the study period, the dose was adjusted in 129 (19.9%) out of the 553 patients. The dose was most frequently modified from 8 mg to 16 mg (84 cases).
Table 1
Patient characteristics (FAS population)
![]()
Efficacy
The mean pain intensity was changed from the NRS value of 5.07 ± 1.99 at baseline to 2.75 ± 1.94 at the final evaluation (Table 2). Therefore, the mean NRS change for pain intensity from baseline to week 4 was 2.32 ± 2.16, indicating a statistically significant decrease in pain intensity (P < 0.001). The % PID was 42.13 ± 46.53, which represented a statistically significant decrease at week 4 as compared to baseline (P < 0.001). The proportion of the patients with % PID ≥ 30 was 70.34% (95% confidence interval [CI], 66.54–74.15) and that with % PID ≥ 50 was 52.08% (95% CI, 47.92–56.24).
Table 2
Change in pain intensity as a primary endpoint (FAS population)

FAS = full analysis set, SD = standard deviation, Min = minimum, Max = maximum, NRS = numerical rating scale.
*P value by Wilcoxon signed rank test; †% PID, Pain intensity difference = (NRS at the baseline − NRS at final visit) / NRS at baseline × 100.
![]()
With regards to the percentage change in pain intensity according to the type of analgesics used previously, the mean NRS change at week 4 compared to baseline was 1.67 ± 3.27 (P = 0.375) in the non-opioid arm, 2.34 ± 2.04 (P < 0.001) in the weak opioid arm, and 2.20 ± 2.32 (P < 0.001) in the strong opioid arm (Table 3).
Table 3
Changes in NRS scores from baseline to final visit by the type of previous analgesics (FAS population)*

FAS = full analysis set, SD = standard deviation, Min = minimum, Max = maximum, NRS = numerical rating scale.
*Missing values were evaluated based on data for early termination; †P value by Wilcoxon signed rank test.
![]()
The degree of pain decreased significantly in patients receiving pain treatment or analgesics over the previous 24 hours (visit 1: 49.60% ± 22.92%, visit 2: 67.14% ± 23.31%, mean change: −17.54% ± 30.22%, P < 0.001) (Table 4). The extent of sleep disturbance due to pain for the past week improved from the NRS value of 3.06 ± 2.64 at visit 1 to 1.46 ± 1.83 at visit 2, indicating a statistically significant decrease (mean difference: 1.61 ± 2.57, 95% CI: 1.39–1.82) at visit 2 as compared to visit 1 (P < 0.001). The degree of interference with performing daily activities due to breakthrough pain for the past week showed a mean NRS change of 1.22 ± 2.30 (from 5.15 ± 2.12 to 3.93 ± 1.83, P < 0.001). The degree of interference with overall pain control caused by end-of-dose failure was also improved from the NRS value of 4.60 ± 1.75 to 3.93 ± 1.70, indicating a statistically significant decrease (P = 0.007).
Table 4
The mean differences of secondary pain assessment (FAS population)*

FAS = full analysis set, SD = standard deviation, NRS = numerical rating scale, CI = confidence interval.
*Missing values were evaluated based on data for early termination; †P value by Wilcoxon signed rank test and two-sided 95% CI on the NRS difference from baseline to final visit.
![]()
In terms of change in the frequency of waking during sleep due to pain between visit 1 and visit 2, it was found that the frequency of waking decreased in 258 subjects (46.7%), did not change in 262 subjects (47.4%), and increased in 33 subjects (6.0%). This indicated that the number of subjects showing a decreased frequency of waking was greater than the number of subjects showing an increased frequency of waking. The state of sleep for the past week was evaluated as good in 55.9% (309/553) of subjects at visit 1 and 87.5% (484/553) of subjects at visit 2, suggesting that the state of sleep at visit 2 was improved as compared to visit 1. The percentage of those experiencing breakthrough pain for the past week was 54.6% (302/553) at visit 1 and 25.7% (142/553) at visit 2, with a statistically significant decrease at visit 2 as compared to baseline (P < 0.001). Out of 132 subjects, the frequency of experience of breakthrough pain for the past week decreased in 76 subjects (57.6%), did not change in 41 (31.1%), and increased in 15 (11.4%). It was found that the percentage of those experiencing increased pain while drug efficacy dropped prior to further treatment with analgesics was 61.6% (175/284) at visit 1 and 17.5% (97/553) at week 4, indicating a statistically significant decrease in experience of end-of-dose failure (P < 0.001). Investigators evaluated the study drug as effective in 72.9% (403/553) of the patients during overall assessment at visit 2, and 72.7% (402/553) of patients were satisfied with the study drug. The most important reason for patient satisfaction was ‘excellent analgesic effect’ (220 patients, 39.8%).
Safety
Of the 678 patients, a total of 76 (11.75%) patients reported adverse events (AEs). The most frequent adverse event was constipation, which occurred in 16 subjects (2.47%) with 16 events, followed by nausea in 15 subjects (2.32%) with 15 events, dizziness in 8 subjects (1.24%) with 8 events, and diarrhea in 7 subjects (1.08%) with 7 events (Table 5). Serious adverse events (SAEs) were reported in 20 patients (3.09%). Treatment-related AEs were reported in 42 (6.49%) patients. The SAE related to the study drug was reported in only one (0.15%) subject, but was manageable. There were 10 deaths in this study, but none of death was related to the study medication.
Table 5
Adverse events with the incidence of ≥ 1% (safety population)

![]()
Among the total of 118 cases of adverse events, mild, moderate, and severe adverse events were reported in 65 (55.08%), 34 (28.81%), and 19 (16.10%) cases, respectively. It was found that the actions taken with regards to the study drug were recorded as ‘no change’ in 59 cases (50.00%), ‘dose changed’ in 17 cases (5.93%), ‘temporarily interrupted’ in 3 cases (2.54%), and ‘permanently discontinued’ in 49 cases (41.53%).
DISCUSSION
Long-acting opioid formulations can improve chronic pain management by providing stable plasma concentrations resulting in around-the-clock analgesia with fewer daily doses (25). OROS hydromorphone is a strong synthetic opioid designed to maintain a constant blood concentration level with once daily dosing (1820). It has been reported to have the advantages of convenience, effectiveness, a good safety profile, a reduction in overall breakthrough pain, and quality of life benefits (82125).
In the current study, cancer patients who required administration of opioid analgesics for pain control received OROS hydromorphone for 4 weeks. Analysis of the primary objective in the FAS group showed a significant decrease in pain intensity. The mean NRS change was 2.32 ± 2.16 and 70.34% of patients experienced ≥ 30% pain intensity improvement. In secondary pain assessments, the degree of sleep disturbance, the degree of breakthrough pain assessed by interference with performing daily activities, and the degree of interference with pain control caused by end-of-dose failure were also significantly improved. These results indicate that symptomatic improvement was seen in pain intensity, sleep disturbance due to pain, breakthrough pain, and end-of-dose failure. In addition, a satisfaction rate of greater than 70% was found for both investigators and subjects.
The changes in pain intensity at week 4 according to the type of analgesics used previously was significant in both of weak opioid and strong opioid arm, not in non-opioid arm (Table 3). In a prospective study of clinical efficacy of OROS hydromorphone, Han et al. (26) also investigated the effect of pain relief according to type of previous used analgesics, such as non-opioid, weak opioid, strong opioid, and adjuvant analgesics (i.e., antidepressants, anticonvulsants, etc.). As similar to present study, decrease in pain intensity score was demonstrated homogenously and statistically significant in all categories except adjuvant analgesics. The difference in scores between visits tended to be diminished from non-opioid to strong opioid. The author of the study commented that the sample size of adjuvant analgesics was too small to establish the statistical significance of the difference. In our study, only six patients were enrolled to non-opioid category and it may be responsible for insignificant change in pain intensity score. However, patients with previous non-opioid use showed a tendency to improvement in pain intensity (mean 1.67 ± 3.27, range −2 to 7). Thus, clinical trials with vigorous enrollment and uniform distribution of subjects are necessary to confirm the efficacy of OROS hydromorphone for cancer pain regardless the type of previously used analgesics.
We further analyzed sleep disturbance and breakthrough pain in patients with baseline cancer pain of moderate to severe intensity. The sleep disturbance rate was significantly decreased from 66.67% (182/273) at visit 1 to 39.19% (107/273) at visit 2 and from 77.40% (113/146) to 44.52% (65/146) in moderate pain and severe pain groups, respectively. The rate of patients experiencing breakthrough pain also significantly decreased from 60.44% (165/273) at visit 1 to 26.37% (72/273) at visit 2 and from 62.33% (91/146) to 34.93% (51/146) in moderate pain and severe pain groups, respectively. But the pain improvement was not influenced by the use of concomitant therapy (2.33 ± 2.18 vs. 2.32 ± 2.14, P = 0.970). These results show that there were improvements of sleep disturbance and breakthrough pain regardless of baseline pain intensity and concomitant therapy.
Comparisons according to efficacy of OROS hydromorphone in recent studies of cancer patients are shown in Table 6. Song et al. (27) reported the efficacy and tolerability of OROS hydromorphone in opioid-naïve cancer patients. They showed favorable outcomes of OROS hydromorphone in % PID as a single and front-line opioid therapy. Considering a short period of time and first experience of opioid in their study, the present study demonstrated comparable data in PI score change and % PID. Han et al. (26) released a prospective study of clinical efficacy of OROS hydromorphone in cancer patients inadequately controlled by other analgesics, and their results were focused on % PID at eight weeks. Comparing with their study, our study showed superior results of % PID within a shorter time in FAS group. Shin et al. (21) presented the clinical usefulness of OROS hydromorphone in improving sleep disturbance. They showed a similar and meaningful change in degree of sleep disturbance decrease compared with present study, as well as in PI score change and breakthrough pain decrease. However, among several latest trials, comprehensive and detailed assessment in efficacy of OROS hydromorphone by variable parameters was performed only in our study recently, and there was not any study investigating the degree of end-of-dose failure.
Table 6
Comparison according to efficacy of OROS hydromorphone in recent studies of cancer patients

| Items | Present study | Song et al. (27) | Han et al. (26) | Shin et al. (21) |
|---|---|---|---|---|
| Study design and Subjects | Multicenter, open label, prospective, observational, single arm study, opioids use previously | Multicenter, open label, prospective, single arm study, opioid-naïve | Multicenter, open label, prospective study, opioids use previously | Multicenter, open label, prospective study, opioids use previously |
| Primary end point | Degree of decrease in PI score at visit 2 | PID at visit 2 | PID at visit 3 | Efficacy in sleep disturbance at visit 3 |
| Duration of assessment, day | 28 | 14 | 29 ± 7 (visit 2), 57 ± 7 (visit 3) | 14 |
| Change in PI score (NRS) | 2.3 ± 2.2 (FAS) |
2.2 ± 2.1 (FAS) 2.6 ± 1.9 (PP) |
5.7 ± 2.1 to 4.5 ± 2.4 (FAS) (visit 2), 4.3 ± 2.6 (visit 3) | 5.2 ± 1.6 to 4.1 ± 1.9 (ITT) |
| PID, % | ≥ 30%; 70.3 (FAS), ≥ 50%; 52.1 (FAS) (mean 42.1 ± 46.5) | ≥ 30%; 68.6 (FAS), 81.4 (PP), ≥ 50%; 51.0 (FAS), 58.6 (PP) | ≥ 30%; 34.6 (visit 2), 39.2 (visit 3) (FAS), 54.9 (visit 2), 65.3 (visit 3) (PP) | - |
| Sleep disturbance decrease (NRS) | 3.1 ± 2.6 to 1.5 ± 1.8 (FAS) (mean 1.7 ± 2.6) | - | 4.4 ± 3.2 to 3.4 ± 2.9 (visit 2), 3.2 ± 3.0 (visit 3) (FAS) | 5.9 ± 1.9 to 4.1 ± 2.5 (ITT), (mean proportion; 34.9%) |
| Breakthrough pain decrease (NRS) | 5.2 ± 2.1 to 3.9 ± 1.8 (FAS) | - | - | 2.63 to 1.53 times |
| End-of-dose failure (NRS) | 4.6 ± 1.8 to 3.9 ± 1.7 (FAS) (mean 0.7 ± 1.8) | - | - | - |
| Assessed as effective by investigator, % | 72.9 (FAS) | 74.0 (FAS), 75.4 (PP) | 61.2 (visit 2), 63.7 (visit 3) (FAS) | 54.1 (PP) |
Data are presented as percentage (range) or mean ± SD (standard deviation).
PI = pain intensity, PID = pain intensity difference, NRS = numerical rating scale, FAS = full analysis set, PP = per-protocol, ITT = intention-to-treat.
![]()
The adverse events occurred in present study were all reported in previous trials (212324). Constipation and nausea were the top-ranked adverse events in most studies, similar to our study. Dizziness and diarrhea showed relatively lower frequency, but did not be missed in lists of adverse events. In addition, vomiting, somnolence and asthenia were frequently reported in those studies. In our study, adverse events were generally associated with the use of strong opioids. Adverse events such as constipation, nausea, dizziness, and diarrhea occurred in 46 (7.83%) patients. One serious adverse event related to the study drug reported was sedation.
Since this study had a single-arm, observational design without a comparator arm, there may be several limitations so caution is required when evaluating efficacy based on assessment before and after administration of the study drug, as any changes seen may not have been caused by administration of the study drug alone. For example, we permit to use short acting opioids for breakthrough pain and adjuvant medications for neuropathic pain. Second, the use of a single assessment at week 4 is another flaw in the study because this may cause recall bias especially in population with poorly controlled cancer pain. Third, adverse events may be under reported because this study was dependent on volunteered reporting, not systematic assessment (28). Finally, caution is required when interpreting the statistical significance of the results since the sample size was not predetermined by a power calculation based on our hypothesis.
In conclusion, OROS hydromorphone is an effective and tolerable agent for cancer patients. It significantly decreases pain intensity, sleep disturbance, breakthrough pain, and end-of-dose failure. Although the effect of OROS hydromorphone tablets on pain control in cancer patients was demonstrated in the present study, randomized clinical trials are still necessary to confirm the efficacy of OROS hydromorphone for cancer pain in the future.
 XML Download
XML Download