

 PDF
PDF ePub
ePub Citation
Citation Print
Print




INTRODUCTION
Juvenile muscular atrophy of the distal upper extremity has been reported mainly in the Asian region (e.g., Japan and India) since 12 cases were reported by Hirayama et al. (1) in 1959. It has been called "Hirayama disease," "benign focal amyotrophy," and "monomelic amytrophy" by various authors to date and is frequently observed among growing men in their teens and twenties. Hirayama disease has the clinical characteristic of invading only the distal portion of the muscles of a unilateral upper extremity with accompanying symptoms of oblique atrophy, cold paralysis, fasciculation, and tremor. Although the pathological and physiological mechanisms are currently unclear, the major cause is anterior shifting of the dural sac, which occurs in cases of neck flexion because it causes an ischemic change in the anterior part of the spinal cord (2). Usually, there is no family history, while some researchers believe that genes such as KIAA1377 and C5ORF42 might be relevant (3). Cases with rare progress deviating from ordinary clinical aspects are occasionally reported, and they typically include bilateral symptoms. In one Japanese study, approximately 3.1% of the patients showed asymmetric invasion in both upper extremities (2). There are even fewer cases of intrusion of the proximal upper extremity.
Our patient showed muscle weakness of proximal upper extremity through secondary progression. We describe a case with atypical progression pattern, and we discuss the clinical meaning for clinicians.
CASE DESCRIPTION
In 15th January 2015, a 29-year-old man visited the hospital with a 1-year history of weakened muscle strength of the proximal portion of the left upper extremity. The patient was diagnosed with Hirayama disease 9 years previously in China. He had a muscle weakness in the distal portion of the left upper extremity, while there was no further progression of the muscle weakness afterward. Although he had weakness, it was not bad enough to be functional. Recently, however, he felt that the functionality and power of the proximal upper extremity was falling. He reported no muscle weakness aggravation during winter, pain, dysesthesia, hypoesthesia, or dysuresia. He also had no extraordinary family history.
The physical examination results indicated the following: blood pressure, 120/70 mmHg; pulse, 77/min; height, 170 cm; and weight, 59 kg. Apparent oblique atrophy was observed in the left forearm except for the brachioradialis muscle and in the hand muscle compared to the right side (Fig. 1A). Moreover, atrophy was observed in the left biceps and deltoid muscles (Fig. 1B). Fasciculation in left hand muscle and irregular tremor of the fingers during finger extension were also observed. Range of motion of the joint was normal, and in an evaluation using the Medical Research Council (MRC) scale for muscle strength, left wrist flexor and extensor muscle strengths and left finger flexor, extensor, and abductor muscle strengths decreased to the 3/5 level, while those of the left elbow flexor and extensor and shoulder abductor decreased to the 4/5 level. The deep tendon reflex was normal, and no pathological reflex was observed.
 | Fig. 1Gross atrophy view of the patient. (A) Distal oblique portion of the left arm. (B) Proximal portion of the left arm.
|
Magnetic resonance imaging (MRI) of the cervical vertebrae in the neutral position showed loss of normal lordosis in the cervical vertebrae but no spinal cord atrophy. MRI of the cervical vertebrae portion in maximum flexion showed neither clearly anterior shifting of the dural sac nor expansion of the epidural venous plexus behind it.
Nerve conduction study findings of bilateral upper and lower extremities were normal. A somatosensory evoked potentials (SEP) test was normal, and no extraordinary changes were observed in cases of maximum flexion of the cervical vertebrae.
In a needle electromyography (EMG) test performed on the left upper extremity, abnormal spontaneous activity was observed in the pronator teres, flexor carpi radialis, flexor carpi ulnaris, and abductor digiti minimi muscles. In all but the brachioradialis muscle, polyphasic motor unit action potentials (MUAP) with increased amplitude and delayed duration were observed. Recruitment type decreased in all of the tested arm muscles (Table 1). No abnormal finding was observed in the bilateral paracervical muscles.
Table 1
Electromyography findings
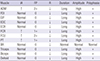
IA, insertional activity; FP, fibrillation potential; R, recruitment; ↑, increased; ↓, reduced; ADM, abductor digiti minimi; FDI, first dorsal interosseous; EIP, extensor indicis proprius; EDC, extensor digitorum communis; ECR, extensor carpi radialis; FCR, flexor carpi radialis; FCU, flexor carpi ulnaris; PT, pronator teres; BR, brachioradialis.
![]()
DISCUSSION
Several case reports have described Hirayama disease, a benign and characteristically slowly progressing disease. Approximately 70% of patients experience disease progression within 3 years, and the disease stagnates after 5 years in approximately 95% of patients (4). Although most known cases involve invasion in a unilateral distal upper extremity, some cases show invasion in the upper extremities of both sides and some show progression within a unilateral proximal upper extremity. In particular, progression within a unilateral proximal upper extremity is very rare, and according to Gourie-Devi et al. (5), only four of 44 (2.3%) patients showed apparent weakness and muscle atrophy in the proximal portion. Moreover, Huang et al. reported that only two of 40 patients had invasion of the biceps (5.0%) and only one had deltoid invasion (6). In another report, a 7-year-old Turkish girl showed weakness in the proximal upper extremity on clinical observation but normal EMG results (7).
To date, no report has examined additional disease progression after 10 years (2). Hence, considering that our patient’s secondary symptom progression occurred a long time after disease onset, we believe that follow-up of the upcoming changes has great significance.
MRI findings showed no abnormal cervical vertebrae, and anterior shifting of the spinal cord during cervical vertebrae flexion, which is the best-known pathophysiological cause of Hirayama disease, was not observed. This is consistent with the report stating that in cases of Hirayama disease, abnormal MRI results of the cervical vertebrae are no longer observed approximately 10 years after the initial occurrence (8). However, the concept that spinal cord damage relevant to cervical vertebrae flexion is the main mechanism underlying Hirayama disease is not fully supported, and the patient may have never had such abnormal findings (9).
Thus, EMG becomes more important for patients diagnosed using medical imaging of insufficient reliability. Unless the EMG tester understands the rare progressive aspect of this disease, confusion can occur when testing the proximal upper extremity muscles. In particular, among benign motor neuron diseases, unilateral brachial amyotrophic diplegia develops from the distal muscle in some cases. According to one report, symptoms of brachial amyotrophic diplegia appeared in the distal muscles, which are controlled by the middle and lower cervical vertebral spinal cord nerves, and progressed toward the proximal muscles, which are controlled by the upper cervical vertebral spinal cord nerves (10). This finding is similar to that of Hirayama disease. As the progressive aspect and treatment plans differ between the two diseases, caution is required when performing differential diagnosis.
On EMG examinations of patients with Hirayama disease, the proximal muscles of the biceps and deltoid, which are controlled by the C5 and C6 nerves, generally appear normal (11). However, needle EMG findings of the biceps and deltoid can produce decreased recruitment and polyphasic MUAP with increased amplitude and long duration, as in this case, although it is very rare. This finding suggests recent disease progression in the proximal portion of the extremity. Similarly, a 23-year-old man in Brazil had monomelic amyotrophy that invaded the unilateral proximal upper extremity and tested malignant on both neurological and EMG screenings (12). In that case, progression to the proximal portion occurred in the early disease stage, unlike the present case, in which invasion into the proximal upper extremity occurred 9 years after disease onset. The SEP test, performed with the neck in a neutral and flexed position, generally shows no abnormal findings; this result was consistent with that observed in the present case (11).
According to Lin et al. (13), Hirayama disease can invade the proximal upper extremity, and overlooking this possibility can result in diagnostic error or confusion. Moreover, MRI findings of cervical vertebra flexion can play a critical role in early diagnosis. However, the present case was different in that proximal invasion occurred several years after disease onset. As we could not rely on the medical imaging findings, background knowledge on the rare progression aspect of this disease and accurate EMG of the proximal upper extremity muscles were critical.
Hirayama disease remains a rarely reported disease with no successful treatment method other than wearing an orthosis that prevents neck flexion. Hence, an accurate early diagnosis is critical, and prevention of further aggravation is important. However, it is difficult to diagnose nontraditional Hirayama disease subtypes because these are not frequently observed in clinics, and diagnostic caution is required to avoid wrong treatment. Benign motor neuron diseases, including Hirayama disease, rarely become serious, and long-term observation is common in most cases. Gourie-Devi et al. (5) followed 44 patients with upper monomelic amyotrophy for an average of 9.7 years (2.5–23 years), and none had newly progressed disease. However, one study reported benign motor neuron disease in a patient with brachial amyotrophic diplegia progressing to amyotrophic lateral sclerosis that invaded the lower extremity and respiratory muscles over time (10). Monitoring this type of progress is important because the clinical aspects can change from benign to malignant. Hence, it is necessary to track the progression of Hirayama disease by carefully observing its long-term development and examining various cases.
Finally, as benign motor neuron diseases generally require long-term follow-up, we expect that cases of rare conditions such as that described here will be informative for clinicians.
 XML Download
XML Download