

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hemolytic uremic syndrome (HUS) is a syndrome characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury (AKI) (1). HUS typically develops in children, is preceded by bloody diarrhea, and responds well to supportive care. Typical HUS is caused by bacterial infection associated with Shiga toxin–producing Escherichia coli (STEC) or other bacteria and is called STEC-HUS (2). However, atypical HUS (aHUS) can develop at any age; 5%-10% of cases do not have prodromal diarrhea and have a poor prognosis (3).
The major pathogenesis of aHUS involves dysregulation of the complement system, such as genetic abnormalities or autoantibodies, which are responsible for 60%-70% of cases (4). Mutation in the complement factor H (CFH) gene is the most frequent cause of aHUS, followed by membrane cofactor protein (MCP), complement factor I (CFI), C3, complement factor B (CFB), thrombomodulin (THBD), and others (3567). Autoantibodies against CFH are detected in 6%-10% of cases of aHUS (8). Recently, complement-independent forms of aHUS, such as mutations in diacylglycerol kinase ɛ (DGKE) and plasminogen (PLG), are reported (9).
Eculizumab, a humanized monoclonal antibody that blocks complement C5 activation and terminal complement component formation, has recently been proven effective against aHUS (101112). It can rescue native kidney function or allow successful kidney transplantation, and may dramatically change the prognosis of this potentially fatal syndrome (11).
aHUS is often misdiagnosed as thrombotic thrombocytopenic purpura (TTP) or STEC-HUS, all of which show common clinical features of microangiopathic hemolytic anemia and thrombocytopenia. However, the pathogenesis and response rate to plasma exchange (PEX) treatment differ between syndromes (131415). Delayed treatment of aHUS can cause death or end-stage renal disease (ESRD) (15). Therefore, a differential diagnosis of aHUS from other forms of thrombotic microangiopathy (TMA) such as TTP and STEC-HUS is very important for its appropriate management.
Since clinical trials of eculizumab with regard to aHUS began (1112), guidelines for aHUS have been developed in Europe for the standardization of management of aHUS (1617). The guidelines accelerated the detection and clinical trials of patients with aHUS (1618). However, in Korea, the diagnosis and management of aHUS have not been studied sufficiently due to the lack of physicians' awareness and the shortage of referral diagnostic laboratories. Hitherto, only 26 patients with genetically confirmed aHUS are reported in Korea (192021). Furthermore, aHUS management in Korea is in the pre-eculizumab era and needs to be improved.
The guidelines offer recommendations for the management of aHUS in Korea. The guidelines' scope includes the diagnosis and treatment of aHUS, with information on investigator networks in Korea. The guidelines were developed by the Korean aHUS Working Group (KHWG), which was organized to study aHUS in October 2015 and is composed of physicians representing the Korean Society of Pediatric Nephrology, the Korean Society of Nephrology, the Korean Society of Hematology, and the Korean Society on Thrombosis and Hemostasis as experts. The guidelines have largely been adopted from the current guidelines due to the lack of evidence concerning the Korean population. The GRADE system (http://www.gradeworkinggroup.org) is used to classify the strength of the recommendations and the quality of the evidence (Table 1).
Table 1
Strength of recommendations and quality of evidence

![]()
DISEASE OVERVIEW
Definition
HUS was earlier divided into diarrhea-positive and diarrhea-negative HUS. The former, also referred to as typical HUS, primarily results from STEC infections, and less frequently from other infections, including Shigella dysenteriae type 1 infection. All other causes of HUS are referred to as aHUS or diarrhea-negative HUS, even though some patients with non-STEC-HUS also present with diarrhea (22).
Currently, the term aHUS applies to a heterogeneous group of diseases that have TMA associated with some degree of AKI (23). TMA syndromes are united by common clinical and pathological features, including microangiopathic hemolytic anemia, thrombocytopenia, organ injury, and vascular damage manifested by microvascular thrombosis (24). Three typical phenotypes of TMAs are STEC-HUS, aHUS, and TTP, another form of TMA caused by a severe deficiency of ADAMTS13 activity.
For aHUS, multiple underlying disease mechanisms are likely involved, including increased complement activation, drugs, non-Shiga toxin infectious agents, cobalamin deficiency, malignancy, transplant, and autoimmune disease (23). On the basis of current diagnostic criteria, aHUS is usually defined to include all types of HUS that are unrelated to Shiga toxins. However, only HUS associated with complement dysregulation may be defined as aHUS as in these guidelines, because complement dysregulation accounts for most non-STEC cases of HUS and complement blockade using eculizumab should be considered in this group of patients (25).
Ongoing research provides improved understanding of the underlying causes and new therapeutic target of HUS. The definition and nomenclature of aHUS need to be redefined based on the underlying pathophysiology and possible treatment options.
Incidence
The incidence of aHUS in the Korean population is not available due to a lack of data. The prevalence of aHUS in Western populations is thought to be approximately 2 per million in adults and 3.3 per million in children younger than 18 years, whereas the prevalence of aHUS in Japan was much lower than that of Western populations and was estimated as approximately 0.84 per million population according to the Nara Medical University Registry (262728). Since April 2012, when aHUS emerged at the global level, 681 patients have been enrolled worldwide by January 30, 2015 (29), and over 1,000 patients with aHUS caused by genetic abnormalities of the complement system are reported (1530313233343536).
Pathogenesis of thrombotic microangiopathy
Complement-mediated aHUS
aHUS is caused by complement dysregulation. An alternative complement pathway is constitutively activated and tightly regulated in normal conditions by multiple regulators to prevent damage to the endothelium and platelets. However, uncontrolled and excessive activation of this pathway, mostly due to genetic mutations or autoantibodies against numerous regulator proteins in the complement system, occurs in patients with aHUS and causes various clinical manifestations (2437). The complement cascade can cause lysis of target cells by forming a pore in the cell membrane. Failure of normal control mechanisms to downregulate the alternative pathway may cause endothelial damage. Complement activation triggers several inflammatory responses. Endothelial cells express complement receptors; they are also susceptible to complement attack. Renal cells are especially sensitive to complement activation, which may explain the predominance of AKI in aHUS. Von Willebrand factor (VWF) may activate the complement (38). Hemostatic factors involved in the clotting cascade, especially those with regulatory roles, interact with complement proteins, but their specific mechanisms and roles are less well understood.
ADAMTS13-mediated TTP
TTP is caused by the severely deficient activity of the ADAMTS13 protease, clinically defined as an activity level < 10% (39). ADAMTS13 cleaves VWF multimers attached to the endothelial surface and prevents the formation of platelet microthrombi. ADAMTS13 deficiency is caused by acquired autoantibodies to ADAMTS13 or hereditary ADAMTS13 gene mutations. The histopathology of TTP and other primary TMAs is characterized by small vessel changes, including swelling of endothelial cells and the subendothelial space, along with vessel wall thickening and platelet microthrombi, typically in small arterioles and capillaries (40). Renal and central nervous system involvement is common.
Other types of TMA
Recently, complement-independent forms of aHUS, such as mutations in DGKE and PLG, have also been reported (924). The implicated factors are generally negative regulators of coagulation or fibrinolysis that also cross-talk with complement factors. Affected individuals generally present during infancy or early childhood. Recessive mutations in the gene encoding DGKE or intronic DGKE mutation (c.888+40A>G) have been reported in familial TMA. Homozygous or compound heterozygous mutations may be seen. DGKE is expressed in endothelium, platelets, and podocytes. Because DGKE is a control enzyme that inactivates the diacylglycerol (DAG) signaling pathway, which promotes thrombosis, loss of DGKE function results in endothelial injury, podocyte dysfunction, thrombosis, AKI, and aHUS (941). Four variants in PLG (c.112 A>G, p.Lys38Glu; c.2134 G>A, p.Gly712Arg; c.758 G>A, p.Arg253His; c.505 C>T, p.Pro169Ser) are associated with aHUS (42). Because plasmin is able to disintegrate formed platelet aggregates, reduced proteolytic activity of plasmin may result in a prethrombotic state of aHUS development (43).
TMA has been reported in individuals with mutations in the gene encoding methylmalonic aciduria and homocystinuria type C (MMACHC) (44). Homozygosity or compound heterozygosity appears to be required for clinical disease. MMACHC is involved in cobalamin (vitamin B12) metabolism. Infants with cobalamin C disease, a type of methylmalonic acidemia, present with various neurologic and developmental findings. The patients show markedly elevated plasma homocysteine levels, but plasma cobalamin levels are normal. Hyperhomocysteinemia-induced damage to glomerular endothelium has been suggested as the putative mechanism for aHUS (45). Complete responses of this TMA to the accessible and inexpensive therapy with high-dose cobalamin and folinic acid have been reported. Evaluation of these abnormalities in cobalamin metabolism is available by measuring serum homocysteine and methylmalonic acid levels.
Drug-induced TMA (DITMA) has been reported following exposure to several types of drugs, especially those containing quinine. Drug-induced antibodies reactive with endothelial cells and possibly margination of granulocytes in renal glomeruli may be responsible for aHUS (46).
Etiology
Defective complement regulation by mutations or autoantibodies is responsible for 60%-70% of cases of aHUS (Fig. 1) (192437). aHUS can occur due to a mutation in one of the several genes encoding complement factors. CFH, CFI, CFB, C3, THBD, and MCP have been implicated.

CFH mutations are the most common abnormality according to registry data from the United States and Europe and account for approximately 23%-27% of the mutations (3147). Along with genetic mutations causing decreased CFH activity, 6%-10% of patients develop anti-CFH antibodies (48). CFH autoantibodies impair the regulatory function of the complement system by inhibiting the binding of CFH to C3b. Some cases have a coexisting complement gene mutation, and large deletion of the CFHR1 and CFHR3 genes are associated with the development of auto anti-CFH-associated aHUS.
MCP gene mutations are responsible for 5%-7% of aHUS cases, whereas the CFI and C3 gene have also been reported to occur at frequencies of 4%-8% and 2%-7%, respectively (3147). Up to 12% of patients with aHUS have combined mutations, usually CFH with CFI or MCP (15). Interestingly, the incidence of anti-CFH antibody is demonstrated to be higher (29.7%) in a recent multicenter cohort study of 51 Korean children with aHUS (Fig. 1) (19).
Heterozygous mutations may be sufficient to cause clinical manifestations. As most genes associated with aHUS showed incomplete penetrance, an additional trigger such as infection, drugs, autoimmune disease, vaccination, pregnancy, cancer, or transplantation is necessary to develop full clinical manifestations of aHUS in a patient with a mutation of these genes (314950515253). Therefore, even in patients with combined genetic mutations, the clinical syndrome may not develop until middle age (53).
Several other disorders such as autoimmune diseases, malignancies, drugs, eclampsia or pregnancy associated HELLP syndrome can also cause clinical and pathological features of HUS and should be considered as possible alternative causes or triggering factors of aHUS. However, some of these disorders are likely to provoke TMA directly in which effective treatment of disorders often leads to complete resolution of TMA (24).
Despite substantial advances in identifying defects in the complement regulation, 35% to 40% of cases of aHUS are still classified as “idiopathic” because no demonstrable genetic mutations have been found (4). Whether these patients have no complement abnormalities or unknown genetic mutations of the complement system remains unknown at present.
Clinical presentations and prognosis
Common symptoms of aHUS include gastrointestinal phenomena such as abdominal pains or diarrhea, although it should be noted that such symptoms may be insufficient to differentiate aHUS from typical HUS. Nonimmunologic microangiopathic hemolytic anemia and thrombocytopenia are the most common clinical manifestations, which ultimately affect the kidneys and brain, leading to acute renal failure and neurologic abnormalities such as confusion or convulsion. The disease may also invade the cardiovascular system, resulting in cardiac failure, myocarditis, and distal gangrene. The most common sequelae resulting from aHUS are chronic renal failure and hypertension (54). Upper respiratory tract symptoms are also commonly observed. aHUS may occur secondarily in patients with malignant hypertension, during pregnancy, after transplantation, with malignant diseases, or while using drugs such as anti-cancer chemotherapy or immunosuppressants (31). aHUS recurs frequently and its prognosis is poor, with death rates as high as 25% and progression to end-stage renal disease in 50% (3). Its rate of recurrence and prognosis vary depending on which particular complement mutation or environmental factors are present.
DIAGNOSIS
Clinical diagnosis
The diagnosis of complement-mediated aHUS is made by excluding other forms of TMA. Therefore, aHUS is suspected in patients with TMA without a secondary cause and ADAMTS13 activity > 10%, without evidence of STEC-HUS (grade 1B).
aHUS is clinically defined by the triad of microangiopathic hemolytic anemia, thrombocytopenia, and AKI, which should not be related to coexisting diseases (12172555). However, STEC infection can trigger an aHUS episode in approximately 1% of patients with a complement mutation (mostly MCP mutation in children) (56), and the alternative complement pathway can be transiently activated during the acute phase of STEC-HUS (5758).
The definitions of microangiopathic hemolytic anemia, thrombocytopenia, and AKI are summarized in Table 2 (25315960). However, regarding the urgency of the diagnosis and management of aHUS, a probable diagnosis of aHUS can be made with two essential criteria of microangiopathic hemolytic anemia and thrombocytopenia, without AKI, in which case the disease should not be related to Shiga toxins or TTP. The diagnostic sensitivity of aHUS would be increased if the data from multiple time points were evaluated, which would help in the early diagnosis and appropriate treatment of aHUS (25).
Table 2
Definitions of micro-angiopathic hemolytic anemia, thrombocytopenia, and AKI
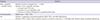
LDH, lactate dehydrogenase; AKI, acute kidney injury; KDIGO, kidney disease improving global outcomes classification; RIFLE, risk, injury, failure, loss, end-stage kidney disease classification; AKIN, acute kidney injury network classification.
![]()
Additionally, aHUS should also be strongly suspected if the following conditions are present in patients with HUS: very young age at onset (< 6 months); time point of HUS onset is not definite; recurrent HUS; recurrent HUS after renal transplantation; familial HUS; no history of diarrhea or bloody stools; and low C3 levels (25).
Distinguishing aHUS from STEC-HUS: Shiga toxin test
STEC-HUS is accompanied by bloody diarrhea due to STEC infection. However, a diagnosis of STEC-HUS should not be made by the diarrhea symptom alone, because diarrhea can also be found in some patients with aHUS and asymptomatic shedding of STEC infection is possible (25). Therefore, tests for Shiga toxin/enterohemorrhagic Escherichia coli (EHEC) to exclude STEC-HUS should be performed for all patients suspected of having aHUS (grade 1B).
Various methods can be used to confirm the presence of Shiga toxins in stool specimens. A stool or rectal swab should be performed at admission, and the specimens should be sent to the laboratory as soon as possible for STEC culture and Shiga toxin testing. If the stool specimens cannot be processed immediately, they should be refrigerated until they can be tested. A stool culture for STEC can be performed using sorbitol MacConkey agar for O157:H7 and selective media for non-O157 STEC (12).
Nonculture assays can be performed to detect Shiga toxins such as STEC immunoassays using commercial kits, whereas real-time polymerase chain reaction (PCR) assays can be used to detect the stx1 and stx2 genes (616263) and serum anti-lipopolysaccharides antibodies against common STEC serogroups (12).
Although sensitivity and specificity of immunoassays approved by the Food and Drug Administration (FDA) for the diagnosis of Shiga toxin producing STEC infection are 92%-100% and 98%-100%, respectively (64656667), one report demonstrated the superiority of PCR over EIA for the detection of STEC (68). It suggested that the level of detection for the EHEC enzyme immunoassays was 106-107 CFU/mL of O157 STEC, but that PCR was able to detect STEC in a suspension of 102 CFU/mL (68).
Distinguishing aHUS from TTP: the ADAMTS13 test
Because the central role of ADAMTS13 protease was discovered in the pathophysiology of acquired TTP (6970), several studies have assessed the clinical applicability of ADAMTS13 activity assay to attempt distinguishing aHUS from TTP (7071). The results of these studies support the hypothesis that a severe deficiency of ADMATS13 activity is more consistent with a diagnosis of acquired TTP than one of aHUS. Currently, in patients without a secondary cause of TMA, ADAMTS13 activity of < 10% is considered to be suggestive of TTP; otherwise, a diagnosis of aHUS is suggested (1217727374). Therefore, tests for ADAMTS13 activity should be performed to exclude TTP in all patients suspected of having aHUS (grade 1B). However, because deficiency of ADAMTS13 activity could also be observed in a small number of patients with aHUS, if clinical findings are highly suggestive of aHUS, the diagnosis of aHUS should not be excluded only based on the result of ADAMTS13 activity test, especially in the patients who do not respond to the plasma exchange (7576).
Currently, three kinds of assays are most widely used for evaluation of ADAMTS13 activity: immunoblotting-based assays, fluorescence resonance energy transfer (FRET)-based assays, and collagen-binding assays (CBA). To avoid erroneous results, the specimen for assaying ADAMTS13 activity should be collected before PEX or plasma infusion using sodium citrate as the anti-coagulant, not EDTA. The specimen should be packaged and shipped to the reference laboratory after freezing to preserve the enzyme activity (73).
Biopsies may be helpful for difficult diagnostic situations. Regardless of whether they are clinically involved, gingiva, skin, and bone marrow are suggested sites for this purpose (77). The thrombi of TTP are typically composed of platelets and VWF with minimal fibrin components. Vascular or perivascular infiltration of inflammatory cells is rare (7879). In contrast, the micro-thrombi of aHUS are dominantly composed of fibrin, and infiltration of inflammatory cells in the perivascular area is usually observed (7980). A recent study suggested that extensive microvascular deposition of C5b-9 supports the diagnosis of aHUS or TTP with concomitant complement dysregulation (81). Therefore, the use of tissue biopsies could be considered on a case-by-case basis along with the ADMATS13 assay.
Distinguishing aHUS from cobalamin defect HUS
Because HUS can develop in hereditary defects in cobalamin metabolism, tests for plasma homocysteine, methionine, and methyl-malonic acid to exclude cobalamin defect HUS are recommended for all patients suspected of having aHUS (grade 2B).
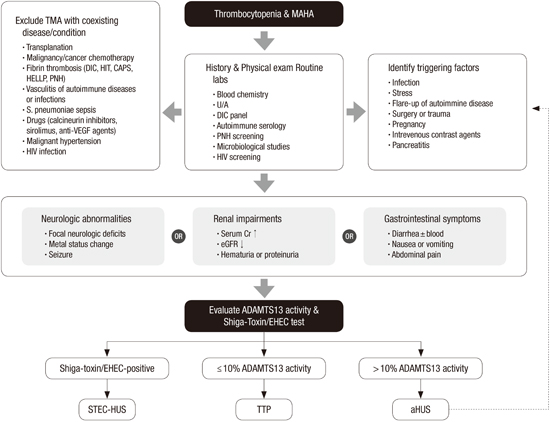
Proposed diagram to differentiate aHUS among TMAs
In patients with thrombocytopenia and MAHA, TMA could be suspected clinically. MAHA could be identified by hemolytic anemia and the presence of schistocytes on the peripheral blood smear. In these patients, a thorough medical history investigation and physical examination should be conducted for a differential diagnosis of TMAs. The first step is to exclude other causative clinical situations of TMA. Examples of TMA with a coexisting disease or condition are summarized in Fig. 2.
 | Fig. 2Proposed diagram to differentiate atypical hemolytic uremic syndrome (aHUS) among thrombotic micro-angiopathies.
MAHA, microangiopathic haemolytic anaemia; TMA, thrombotic microangiopathy; DIC, disseminated intravascular coagulation; HIT, heparin-induced thrombocytopenia; CAPS, Catastrophic antiphospholipid syndrome; HELLP, hemolysis, elevated liver enzymes, and low platelet count syndrome; PNH, paroxysmal nocturnal hemoglobinuria; VEGF, vascular endothelium growth factor; U/A, urine analysis; HIV, human immunodeficiency virus; ADAMTS, A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13; EHEC, enterohemorrhagic Escherichia coli; STEC, Shiga toxin-producing Escherichia coli; HUS, hemolytic uremic symdrome; TTP, thrombotic thrombocytopenic purpura; aHUS, atypical hemolytic uremic symdrome.
|
Because TMA is a multisystem disease, the presence of any organ injury should be identified by physical examination and laboratory studies. The nervous, renal, and gastrointestinal systems are most frequently involved in patients with TMAs. First, the physical examination should be focused on neurological or gastrointestinal abnormalities. Previously, neurological abnormalities have been used to differentiate TTP from other TMAs, because the involvement of the nervous system was reported in up to 79% of patients with TTP (8283848586). However, various degrees of neurological injury have also been observed in ADAMTS13 nondeficient aHUS patients (3878889). Therefore, the clinical utility of neurological symptoms is somewhat limited. Similarly, unexplained accompanying gastrointestinal symptoms are commonly observed in patients with TMAs (390). Nevertheless, it is noteworthy that patients with bloody diarrhea should be examined for Shiga toxin–producing bacteria. Although renal injury requiring dialysis is the more frequent manifestation among aHUS patients, various degrees of renal impairment are also observed in patients with TTP (828491). Other laboratory tests for rare conditions associated with TMAs may be helpful in differentiating the causes of TMAs. These include disseminated intravascular coagulation (DIC) panel (PT, aPTT, fibrinogen, FDP, d-dimer, and others), autoimmune serology (FANA, ANCA), paroxysmal nocturnal hemoglobinuria (PNH) screening by flow cytometry, and human immunodeficiency virus (HIV) screening. Therefore, the following laboratory tests for rare conditions associated with TMAs may be helpful in differentiating the causes of TMAs: DIC panel, autoimmune serology, PNH screening by flow cytometry, and HIV screening (grade 1B).
In patients with TMA and any organ injury of the nervous, renal, or gastrointestinal system, an assay for ADAMTS13 activity and Shiga toxin/EHEC should be performed (grade 1B). Patients positive for Shiga toxin or EHEC have STEC-HUS diagnosed. Patients with severely deficient ADAMTS13 activity of ≤ 10% have TTP diagnosed; otherwise, (> 10% of ADAMTS13 activity) aHUS is diagnosed. Consequently, genetic screening for complementary abnormalities should be performed for aHUS patients (grade 1B).
In patients diagnosed with aHUS, the possibility of triggering factors should be investigated. Even in patients with a dysregulated complement system, the clinical syndrome may not develop until middle age (3). Certain clinical situations may promote the activation of the complement system and, consequently, the clinical manifestation of aHUS may develop in unaffected healthy carriers. Typical examples include infection, stress, active inflammation, surgery, pregnancy, and intravenous contrast agents (74). Any identified triggering factor should be eradicated as soon as possible. To prevent recurrent episodes, patients with aHUS should be educated to avoid the identified triggering factors as much as possible after achieving clinical remission (grade 1C).
Further investigation: genetic screening
Abnormalities of the complement regulation system are the main causes of aHUS and include various kinds of mutations and copy number variations in the genes encoding C3, CFH, CFI, CD46 (MCP), CFB, THBD, and complement factor H–related proteins 1 through 5 (CFHR1-5) or anti-CFH antibodies (1292). Currently, 60%-70% of patients with aHUS have identifiable mutations in complement genes or anti-CFH antibodies (493).
First, serum C3 and C4 levels should be measured for all patients presenting with clinical features of HUS. If those levels have decreased, then aHUS should be suspected, although the C3 and C4 levels may be normal in aHUS. If possible, one should measure factor H and factor I levels in serum and CD46 expression in PBMCs using FACS, if possible, for all patients presenting clinical features compatible with a diagnosis of aHUS, because the results guide the prognosis and transplantation options (121792). Similarly, however, normal levels or expression of factor H, factor I, and MCP do not rule out the possibility of a normally expressed but functionally impaired mutant. Therefore, screening for complementary abnormalities by measuring serum levels (C3, C4, CFH, CFI, CFB, anti-CFH antibody) and expression of MCP in peripheral blood mononuclear cells using flow cytometry should be performed for aHUS patients (grade 2B).
Eventually, the full genetic analysis is recommended for all patients with aHUS who show no causative disease, STEC infection, severe ADAMTS 13 deficiency, or hyperhomocysteinemia/methyl-malonic aciduria (grade 1B) (12). Additionally, genetic screening should be performed without delay if the patients have an HUS relapse, a familial history of nonsynchronous HUS, pregnancy/postpartum HUS, or de novo post-transplant HUS (12). Genetic screening is especially essential before renal transplantation for aHUS. However, genetic screening is not justified before transplantation for STEC-HUS, unless this diagnosis is uncertain or unproven (12).
There are several reasons for performing a genetic analysis when the first episode of aHUS occurs. First, through genetic analysis, we can confirm whether aHUS is complement-dependent, which might predict prognosis, the risk of relapses, and progression to ESRD according to the results of genetic abnormalities and might guide the complement blockade treatment. Second, we can provide genetic counseling to parents and family. Third, we can make decisions for various renal transplantation situations, such as the choice of donor, treatment guidance for the prevention or treatment of post-transplant recurrence, and whether to opt for combined kidney and liver transplantation (12).
For genetic analysis, screening for mutations in CFH, CFI, MCP, C3, CFB, THBD, and DGKE should be performed by direct Sanger sequencing analysis or next-generation sequencing (NGS), and screening for the CFH hybrid gene and copy number variation in CFH and CFHRs should be performed by multiplex ligation-dependent probe amplification (MLPA) (17). Additionally, because 6%-10% of aHUS patients have antibodies that bind to the C terminal region of factor H, and because the prevalence of anti-CFH in Korean children is higher than in other countries (819), autoantibodies against factor H should be performed in an appropriately accredited laboratory (94).
MANAGEMENT
Supportive treatment
Any patient suspected of having aHUS should be transferred to a specialized center where dialysis and PEX facilities are available. Platelet transfusion is contraindicated unless the patient is bleeding or when a surgical procedure carrying a risk of bleeding is necessary. The protection of peripheral and central veins is important in patients with aHUS who need long-term vascular access for hemodialysis or PEX. Infections can trigger the relapse of aHUS, and appropriate treatment is necessary (92).
Plasma therapy
All patients who are clinically suspected of having aHUS should be offered a trial of PEX and/or plasma infusion (PI) as early as possible (173192). Since the laboratory findings including complement and ADAMTS13 are usually not available when the patient presents with clinical symptoms of aHUS, the guidelines recommend plasma therapy to be started within 24 hours of presentations including renal insufficiency, unexplained thrombocytopenia and a microangiopathic hemolytic anemia with a normal international normalized ratio and partial thromboplastin time. PEX is an alternative option if eculizumab is not available, with an exchange of 1.5 plasma volumes (60–75 mL/kg) per session and with fresh-frozen plasma (FFP) for compensation. When PEX cannot be performed, PI (10–20 mL/kg) should be administered. Patients receive PEX or PI daily from 5 days up to 2 weeks during the acute phase until the platelet count, lactate dehydrogenase (LDH), and hemoglobin levels have normalized and the renal function shows signs of improvement. When disease activity is controlled by daily PEX, the subsequent frequency is 5 times per week for 2 weeks, and then 3 times per week for the subsequent 2 weeks (92). The frequency may be reduced from weekly to every 2 to 4 weeks as a long-term maintenance therapy. A Recent study suggested that the risk of ESRD after the first episode of aHUS was similar in the group with low-intensity plasma therapy and those with the high-intensity plasma therapy. Although the intensity of plasma therapy remains questioned, there is a clinical need for a more efficient treatment in patients with aHUS because of severe renal outcomes (56). The further frequency of plasma therapy has to be decided according to the genetic information and clinical response to plasma therapy. In patients with an MCP/CD46 mutation, the withdrawal of plasma therapy is possible because MCP is not a circulating protein. An attempt to withdraw plasma therapy can be considered in patients without a relapse of HUS, despite tapering of PEX/PI to monthly administration (92). Although PEX/PI induced remission in 55%-80% of episodes in patients with aHUS in the Italian cohort, 48% of children and 67% of adults died or progressed to ESRD during the 3-year follow-up period (31).
Kidney transplantation
The risk of HUS recurrence after kidney transplantation varies according to the underlying genetic abnormality. Renal transplantation alone is not recommended for patients with a CFH or CFI mutation because of the poor outcome, with 80% of patients losing their graft due to a recurrence of the disease within 2 years. In patients with an MCP/CD46 mutation alone, the risk of recurrence post-transplantation is low. Patients with a C3 or CFB mutation show a significant risk of disease recurrence post-transplantation. No post-transplantation recurrence has been observed to date in patients with a DGKE mutation. Patients with an anti-factor H autoantibody should be treated with a PEX combination with Rituximab to minimize the antibody titer before proceeding to renal transplantation (17). Living-related renal transplantation alone should be avoided in cases of aHUS (121792). PEX/PI for post-transplant recurrence usually failed to prevent graft loss, and prophylactic PEX/PI was recommended. According to the Consensus Study Group, one PEX with FFP (60–75 mL/kg) should be performed within 4-6 hours before graft reperfusion, FFP (10–20 mL/kg) should be infused during surgery, and PEX with FFP (60–75 mL/kg) should be continued daily for at least 5 days, followed by 5 sessions per week for 2 weeks, and then 3 sessions per week for 2 weeks, after which it should be tapered on a case-by-case basis. Recently, prophylactic eculizumab treatment for patients at high risk for post-transplant recurrence has been considered (95).
Recommendations
Patients should be informed that the risk of disease recurrence after renal transplantation varies according to the causative mutations. Patients with an anti-factor H autoantibody should be treated with a PEX combination with Rituximab to minimize the antibody titer before proceeding to renal transplantation. Living-related renal transplantation alone should be avoided in cases of aHUS (grade 1C).
Liver transplantation
Because CFH, CFI, CFB, and C3 are synthesized in the liver, an isolated liver transplantation or a combined liver and kidney transplantation may be an option for patients with preserved eGFR, despite a severe and/or relapsing course, and for patients with a CFH, CFI, CFB, or C3 mutation, complications, or no benefit from PEX/PI and no access to eculizumab treatment (12179295).
Terminal complement blockade (eculizumab)
Eculizumab has been used successfully in patients with aHUS since 2009 (96), and it received approval for the treatment of aHUS in the United States and Europe in late 2011. Eculizumab reduces terminal complement activation (C5b-9, membrane attack complex). Eculizumab therapy for patients with aHUS also reduces inflammation, endothelial damage, thrombosis, and renal injury. Therefore, eculizumab therapy reduces the ongoing risk of systemic TMA and progression to organ damage in patients with aHUS (97).
Eculizumab therapy for aHUS is usually delayed for several days because it takes several days to obtain baseline ADAMTS13 activity results, which are necessary for the differential diagnosis between aHUS and TTP. However, eculizumab therapy should be considered as a first-line therapy for patients who already have had aHUS diagnosed or who have had mutation results for aHUS (72). Because TTP is less common in children, and because plasma exchange therapy is ineffective in patients with aHUS, it is also important to start eculizumab therapy as soon as possible in pediatric patients who are suspected of having aHUS (12).
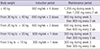
Because eculizumab therapy increases the risk of meningococcal infections, patients should receive a meningococcal vaccination at least 2 weeks prior to receiving the first dose of eculizumab. However, eculizumab therapy usually cannot be delayed in patients with aHUS. Therefore, additional prophylactic antibiotics for 2 weeks (ciprofloxacin 500 mg twice daily, rifampin 600 mg twice daily, or penicillin VK 250 mg four times daily) should be considered for patients with aHUS who are beginning eculizumab therapy (72). Eculizumab therapy is usually initiated intravenously at a dose of 900 mg weekly for the first 4 weeks, followed by 1,200 mg for the fifth dose 1 week later, and then 1,200 mg every other week thereafter. Dose adjustments should be considered for pediatric patients with body weight < 40 kg (1272). The recommended dosage and schedule are summarized in Table 3 (12).
Table 3
Recommended dosage and schedule of eculizumab in patients with aHUS
![]()
Whereas improvement of thrombocytopenia and elevated LDH occur more quickly after eculizumab therapy, recovery of renal function and other end-organ injuries present may take longer. Therefore, recovery from thrombocytopenia is used as a surrogate marker for activity of complement-mediated TMA (10). With continuous therapy, a time-dependent improvement of the estimated GFR is usually observed. Some patients eventually discontinued their previous dialysis. Patients with aHUS who undergo eculizumab therapy have an approximately 50% lower risk of reaching ESRD within 3 months of an aHUS episode compared with historical controls (98). Life-long eculizumab maintenance therapy is usually required for aHUS (10). If aHUS patients discontinue eculizumab treatment, then they should be monitored closely for signs and symptoms of severe complications (99). Monitoring of the complement function test may allow a safe reduction of eculizumab therapy when patients remain in sustained remission (100). However, a continuation of eculizumab therapy is recommended for kidney transplant recipients with CFH mutations, for anti-CFH antibody–positive patients with an antibody titer < 2.5-times the upper limit of normal, and for patients with GFR < 20 mL/min/1.73 m2 (101). Because of another acute episode of aHUS after any complement-activating clinical event (infection, surgery, or pregnancy), careful monitoring and education should be emphasized for patients who have discontinued eculizumab.
Recommendations for the diagnosis and management of aHUS
The Korean aHUS Working Group suggested the recommendations for the diagnosis and management of aHUS (Table 4). Although the guidelines are developed based on the current evidence, there are still many limitations to apply each recommendation in real clinical practice because of limited resources in Korea. The guidelines are not intended to limit, but improve current practice on the management of aHUS. Therefore, these guidelines intend to provide Korean physicians on the general information only and do not replace professional medical practice.
Table 4
Recommendations for the diagnosis and management of atypical hemolytic uremic syndrome

aHUS, atypical hemolytic uremic syndrome; CFB, complement factor B; CFH, complement factor H; CFI, complement factor I; DIC, disseminated intravascular coagulation; EHEC, enterohemorrhagic Escherichia coli; HIV, human immunodeficiency virus; MCP, membrane cofactor protein; PEX, plasma exchange; PNH, paroxysmal nocturnal hemoglobinuria; STEC-HUS, Shiga toxin–producing Escherichia coli hemolytic uremic syndrome; TMA, thrombotic micro-angiopathy.
![]()
 XML Download
XML Download