

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Adenosine is well known to have a cell protective effect in stressful situations, such as ischemia, hypoxia, and inflammation (1). It is released from metabolically active cells into the extracellular space and is produced by the breakdown of released adenosine triphosphate (ATP). While the concentration of extracellular adenosine is about 300 nM in normal cells (2), its concentrations rapidly exceed 1 μM in cells undergoing damage (3). Adenosine’s regulation of tissue function is mediated through activation of a family of 4 G-protein coupled receptors, which includes the A1, A2a, A2b, and A3 adenosine receptors (AR). Concentrations of extracellular adenosine vary widely depending on external factors and the degree of stress, tissue types, and activation levels of the 4 adenosine receptors (4). Previous studies investigating the role of adenosine have focused on the agonists and antagonists of adenosine receptors instead of adenosine itself, because of the limitation of its very short half-life (5).
The A3 adenosine receptor (A3AR) has been detected in a wide variety of tissues. A3AR was highly expressed in the testis, but was less likely to be expressed in the lungs, kidney, heart, and central nervous system. Interestingly, inflammatory and cancer cells possess higher A3AR content than other adenosine receptor subtypes. A3AR is able to play a different roles under ischemic conditions, where it shows pro/anti-inflammatory, cellular proliferation/death, and pro/antitumoral effects depending on different pathophysiological environments (6). Previous studies demonstrated that inhibition of A3AR leads to kidney protection in ischemia-reperfusion injury (7) and myoglobinuria-induced injury (8). A recent study reported that A3AR antagonist ameliorates the progression of tubulointerstitial fibrosis in a unilateral ureteral obstruction (UUO) mice model (9).
Few reports have described the expression and distribution of adenosine receptor subtypes in the kidney and the role of A3AR and A3AR antagonists in kidney injury. A new A3AR antagonist (LJ1888) may be a powerful, highly selective, species-independent, and orally available agent (10). However, there is rare data for the role of A3AR in glomerular kidney injury. The aim of this study was to investigate the mechanism and renoprotective effects of the highly selective A3AR antagonist (LJ1888) in adriamycin (ADX)-induced nephropathy.
MATERIALS AND METHODS
Experimental animals
All 6-week-old Balb/c male mice weighing 22-24 g were purchased from RaonBio Co. (Yongin, Gyeonggi-do, Korea). They were randomly assigned to three groups: control (n = 5); ADX injection (n = 10); or administration of LJ1888 after ADX injection (n = 10). Mice received a single tail vein injection of ADX diluted with 0.9% saline to a final dose of 11 mg/kg. Control mice received the same volume of isotonic saline. Four weeks after ADX injection, mice drank water mixed with 10 mg/kg per day of LJ1888 (Lee et al. [9] College of Pharmacy, Ewha Womans University, Seoul, Korea) for two weeks as previously described. All mice were provided with a standard diet and water, and were maintained at constant temperature (23°C ± 2°C) and humidity (55% ± 5%) with a 12-hour light/dark cycle. Daily food intake was monitored at regular intervals to confirm drug administration. Plasma creatinine level was determined using high-performance liquid chromatography. To determine urinary protein, albumin, and nephrin excretion, mice were caged individually and a 24-hour urine sample was collected at baseline, two, four and six weeks. Urinary albumin concentrations were determined with a competitive enzyme linked immunosorbent assay (ELISA) kit (ALPCO, Westlake, OH, USA). Urinary protein concentrations were determined by colorimetric detection using bicinchoninic acid (Pierce BCA protein Assay Kit, Pierce Biotech. Inc., Rockford, IL, USA). Urinary nephrin measurement was performed with the commercially available ELISA kit (Exocell, Philadelphia, PA, USA). The urinary 8-isoprostane level was measured using an ELISA kit (Cayman Chemical, Ann Arbor, MI, USA). The extent of peroxidative reaction in the kidney tissue was determined by direct measurement of lipid hydroperoxides (LPO) using an LPO assay kit (Cayman Chemical), as described previously (11). Mice were sacrificed under anesthesia with intraperitoneal injections of sodium pentobarbital (50 mg/kg). Their heart, epididymal fat, liver, and kidney tissues were extracted and subsequently snap-frozen in liquid nitrogen.
Analysis of gene expression by real-time quantitative polymerase chain reaction
We extracted total RNA from experimental cells using the Trizol reagent. Primers were designed for their respective gene sequences using the Primer 3 software, and the secondary structures of the templates were examined and excluded using the mfold program (Supplementary Table 1). Quantitative gene expression was performed using a Light Cycler 1.5 system (Roche Diagnostics Corporation, Indianapolis, IN, USA) using SYBR Green technology. In 96-well real-time polymerase chain reaction (PCR) plates, 10 μL SYBR Green master mix was added to 1 μL of RNA (corresponding to 50 ng of total RNA) and 900 nM of forward and reverse primers for a total reaction volume of 20 μL. Real-time reverse transcriptase PCR was performed for 10 minutes at 50°C and for 5 minutes at 95°C. Thirty PCR cycles of denaturation for 10 seconds at 95°C and annealing with extension for 30 seconds at 60°C were then conducted. Gene level ratios relative to β-actin (relative gene expression number) were calculated by subtracting the threshold cycle number of the target gene from that of β-actin and squaring this difference. We evaluated the specificity of each PCR product using melting curve analysis, followed by agarose gel electrophoresis.
Protein extraction and western blot analysis
Nuclear and cytoplasmic proteins were extracted from renal cortical tissues and cells using a commercial nuclear extraction kit according to the manufacturer’s instructions (Active Motif, Carlsbad, CA, USA). Protein concentration was determined using the bicinchoninic acid method (Pierce Pharmaceuticals). For western blotting, 40 μg protein was electrophoresed on a 10% SDS-PAGE mini-gel. Proteins were transferred onto a polyvinylidene difluoride membrane, and the membrane was hybridized in blocking buffer overnight at 4°C with mouse monoclonal anti-nuclear factor (NF)-κB p65 antibody (1:1,000; Cell Signaling Technology, Danvers, MA, USA), rabbit polyclonal anti-transforming growth factor (TGF)-β1 antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal anti-nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (NOX4) antibody (1:500; Novus Biologicals, Littleton, CO, USA) and goat polyclonal anti-toll-like receptor 4 (TLR4) antibody (1:500; Santa Cruz Biotechnology). The membrane was subsequently incubated with horseradish peroxidase-conjugated secondary antibody (1:1,000 dilution) for 60 minutes at room temperature. We detected the specific signals using an enhanced chemiluminescence method (Amersham, Buckinghamshire, UK).
Histological examination and immunohistochemistry
Kidney samples were fixed in 10% buffered formalin and embedded in paraffin. Kidney tissue was cut into 4-µm-thick slices and stained with periodic acid Schiff (PAS), Masson’s trichrome, and sirius red staining. Macrophage infiltration was detected by rat anti-mouse F4/80 antibody (1:2,000; Serotec, Raleigh, NC, USA) and incubated at room temperature for 1 hour, followed by use of the Envision kit (Dako, Carpinteria, CA, USA). To perform immunohistochemical staining for type I collagen, TGFβ1, and α-smooth muscle actin (SMA), kidney sections were transferred to a 10 mM/L citrate buffer solution adjusted to a pH of 6.0. Thereafter, sections were microwaved for 10–20 minutes to retrieve antigens for TGFβ1 and α-SMA, or treated with trypsin (Sigma, St. Louis, MO, USA) for 30 minutes at 37°C for type I collagen and type IV collagen. To block endogenous peroxidase activity, 3.0% H2O2 in methanol was applied for 20 minutes, followed by incubation at room temperature for 60 minutes with 3% BSA/3% normal goat serum. Slides were incubated overnight at 4°C with rabbit polyclonal anti-TGF-β1 antibody and rabbit polyclonal anti-type I collagen antibody (1:200; Santa Cruz Biotechnology), rabbit polyclonal anti-type IV collagen antibody (1:200; Santa Cruz Biotechnology), and rabbit polyclonal anti-α-SMA antibody (1:100; Santa Cruz Biotechnology). For coloration, slides were incubated at room temperature with a mixture of 0.05% 3,3´-diaminobenzidine containing 0.01% H2O2 and then counterstained with Mayer’s hematoxylin. Negative control sections were stained under identical conditions with a buffer solution that was substituted for the primary antibody. A pathologist carried out the histological examinations in a blinded manner.
Statistical analysis
Nonparametric analysis was used because of the small sample size. Results are expressed as mean ± s.e.m. Multiple comparisons were carried out using Wilcoxon’s rank-sum tests and the Bonferroni correction. A Kruskal–Wallis test was used to compare more than two groups, followed by a Mann–Whitney U-test, using a microcomputer-assisted program called SPSS for Windows 20.0 (IBM SPSS statistics, Chicago, IL, USA). P value < 0.05 was considered statistically significant.
RESULTS
Physical and biochemical parameters of experimental animals
The physical and biochemical parameters for each group of the experimental animals are shown in Table 1. Injection of high dose ADX induced dehydration and cachexia in a previous animal study (12). In our study, ADX administration inhibited weight gain. Mice with ADX-induced nephropathy displayed a significant decrease in body weight compared to the control mice four weeks after an 11 mg/kg injection of ADX. At the time of sacrifice, mice in the ADX injection group had significantly lower body weight than mice in the control group, and LJ1888 administration led to recovery of body weight lost during ADX treatment. The body weight of LJ1888-treated mice was significantly increased compared to that of ADX-injected mice at six weeks, despite no significant changes in daily water and food intake. Although urine volume tended to increase at two weeks and decrease at four weeks after ADX injection compared with control mice, there was no significant difference based on LJ1888 treatment. Kidney weight was significantly lower in ADX-induced nephropathy than in the control group. However, LJ1888 treatment did not induce any significant change in kidney weight. ADX increased the plasma creatinine level significantly, which was improved by LJ1888 treatment.
Table 1
Physical and biochemical parameters of experimental animals

Effects of LJ1888 administration on urinary excretion of protein and albumin in ADX-induced nephropathy
ADX leads to direct toxic injury of the glomerular filtration barrier including podocytes, the glomerular basement membrane and glomerular endothelial cells. Podocyte damage induces loss of protein and nephrin in the urine (13). In order to investigate the effects of LJ1888 on podocyte injury in ADX-induced nephropathy, we checked the urinary excretion of protein and albumin. Urinary protein excretion was significantly increased at four and six weeks after ADX injection compared with the control group. LJ1888 treatment inhibited urinary protein excretion during two weeks of treatment in ADX-induced nephropathy (Fig. 1A). In addition, urinary albumin excretion significantly increased four and six weeks after ADX injection compared with the control group, and significantly decreased after two weeks of LJ1888 treatment in ADX-induced nephropathy (Fig. 1B).
Fig. 1
Effects of LJ1888 on urinary excretion of protein and albumin in experimental ADX-induced nephropathy. (A) Twenty-four hour urinary protein excretion level. (B) Urinary excretion of albumin level. Urinary protein, albumin, and nephrin levels were corrected for urine creatinine levels.
Values are expressed as (means ± SEM).
ADX, adriamycin.
*P < 0.01; †P < 0.05 vs. control; ‡P < 0.001 vs. ADX.

Effects of LJ1888 on oxidative stress and urinary nephrin excretion in ADX-induced nephropathy
The mechanism of ADX-induced nephropathy occurs in part through podocyte injury via oxidative stress. ADX exacerbates cellular damage in the renal cortex, inducing renal fibrosis (14). Examination of urinary 8-isoprostane excretion and kidney lipid peroxidation (LPO) concentration is an approach for diagnostic assessment of oxidative stress in the kidney (15). In our study, the urinary level of 8-isoprostane markedly increased six weeks after ADX injection compared with the control group, and significantly decreased after two weeks of LJ1888 treatment (Fig. 2A). Likewise, LPO concentration in the kidney was significantly increased six weeks after ADX injection compared with the control group, but significantly decreased after LJ1888 treatment (Fig. 2B). In addition, we measured urinary nephrin excretion to determine podocyte injury. As shown in Fig. 2C, each group showed no significant difference in urinary nephrin excretion during four weeks after ADX administration. However, nephrin was markedly increased at six weeks after ADX injection compared with the control group. Interestingly, LJ1888 treatment yielded the greatest inhibitory effects on urinary nephrin excretion.
Fig. 2
Effects of LJ1888 on oxidative stress in ADX-induced nephropathy. (A) Twenty-four hour urinary level of 8-isoprostane. Urinary 8-isoprostane levels were corrected for urine creatinine levels. (B) LPO concentration in the kidney tissue. LPO concentration levels were corrected for kidney weight. (C) Urinary excretion of nephrin.
Values are expressed as (means ± SEM).
ADX, adriamycin; LPO, lipoperoxidase.
*P < 0.01 vs. control; †P < 0.001; ‡P < 0.01 vs. ADX.

Effects of LJ1888 on proinflammatory and profibrotic mechanisms in ADX-induced nephropathy
To determine whether LJ1888 has a protective effect in ADX nephropathy, we next investigated the mRNA expression of proinflammatory and profibrotic molecules in renal cortical tissue. Gene expression of profibrotic cytokines including type IV collagen, plasminogen activator inhibitor (PAI)-1, macrophage/monocyte chemoattractant protein (MCP)-1, and NOX4 were remarkably increased in ADX-injected mice. On the other hand, LJ1888 treatment significantly suppressed the expression of PAI-1, MCP-1, and NOX4. TGF-β1 mRNA levels in the renal cortex (Fig. 3A). Similarly, proinflammatory cytokines such as TLR4, tumor necrosis factor (TNFα), interleukin (IL)-1β, and interferon (IFN)-γ were highly expressed after ADX administration compared to the control group, and their mRNA expression was significantly suppressed after LJ1888 treatment. As expected, A3AR mRNA levels significantly increased after ADX injection, but did not change significantly after LJ1888 administration (Fig. 3B). Moreover, western blot analysis confirmed that the renal cortical expression of NF-κB, TGF-β1, and NOX4 were all markedly induced in ADX-injected mice. On the other hand, LJ1888 significantly reduced the expression of all these proteins. TLR4 did not differ among the three groups in the kidney (Fig. 3C).
Fig. 3
Effects of LJ1888 on inflammation and fibrosis in ADX-induced nephropathy. (A and B) Effects of LJ1888 on the expression of inflammatory and fibrotic cytokines. (C) Representative western blots of NF-κB, TGF-β1, NOX4, and TLR4 in the kidney and quantitative analysis scoring of western blot.
Values are expressed as (means ± SEM).
ADX, adriamycin; Col-4, type IV collagen; PAI-1, plasminogen activator inhibitor-1; MCP-1, macrophage/monocyte chemoattractant peptide-1; TGF-β1, transforming growth factor-β1; NOX-4, NADPH oxidase 4; TLR-4,Toll-like receptor 4; A3AR, A3 adenosine receptor; TNFα, tumor necrosis α; IL-1β, interleukine-1β; IFN-γ, interferon-γ; NF-κB, nuclear factor-κB.
*P < 0.05; †P < 0.01 vs. control; ‡P < 0.05; §P < 0.001; ∥P < 0.01 vs. ADX.

Effects of LJ1888 on histomorphological changes in ADX-induced nephropathy
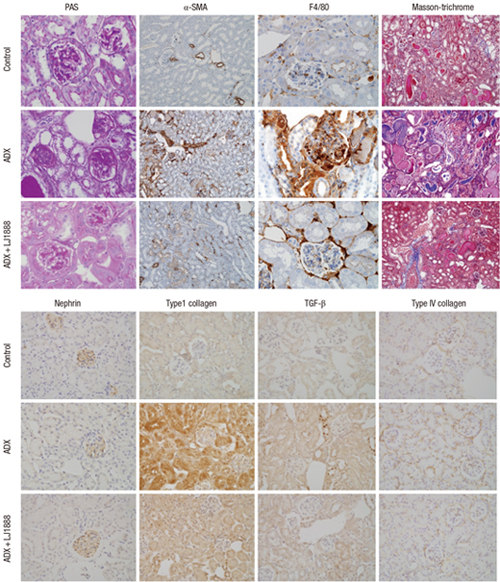
Fig. 4 shows representative renal pathology and immunohistochemical staining for each group of experimental animals at the time of sacrifice. PAS and Masson’s trichrome staining demonstrated that glomerular and tubulointerstitial damage including glomerulosclerosis, excessive collagen deposition, interstitial fibrosis, and tubular atrophy were dramatically increased in ADX-induced nephropathy. The glomeruli had much more severe pathological changes than the tubulointerstitium in ADX-induced nephropathy. ADX led to development of progressive glomerulosclerosis and demonstrated increased accumulation of type I collagen, type IV collagen, TGF-β1, and α-SMA in the glomeruli. In contrast, LJ1888 administration significantly ameliorated severe damage lesions of glomerulosclerosis. Infiltration of inflammatory cells such as macrophages and T lymphocytes in the renal glomeruli is an important pathological finding in the early progression of ADX-induced nephropathy (12). F4/80 is macrophage specific to the inflammatory process. F4/80 staining demonstrated that macrophage infiltration in the glomeruli was markedly increased after ADX injection and dramatically recovered with LJ1888 treatment. Nephrin has important effects in controlling the integrity and functions of the podocyte slit diaphragm structure (16). In a previous study, ADX induced direct toxic damage to the podocytes and loss of nephrin in urine (13). Immunohistochemical staining revealed that the numbers of nephrin-positive cells were dramatically decreased in the glomeruli of ADX-injected mice and were significantly restored by LJ1888 treatment. As shown in Fig. 5, quantitative analysis with immunohistochemical staining demonstrated those results.
Fig. 4
Representative renal histological and immunohistochemical staining in experimental models at the time of sacrifice.
Representative sections show that the histological changes and immunohistochemical staining for PAS, F4/80, Masson’s trichrome and α-SMA, nephrin, type I collagen, TGF-β1, and type IV collagen are increased in ADX-induced nephropathy and decreased after treatment with LJ1888. Original magnification X400.
ADX, adriamycin; PAS, periodic acid-Schiff; α-SMA, α-smooth muscle actin; TGF-β1, transforming growth factor-β1.

Fig. 5
Quantitation of immunohistochemical staining. (A) Glomerulosclerosis index, (B) F4/80 positive cell staining score, (C) Tubulointerstitial fibrosis index, (D) Quantitation of immunohistochemical staining of nephrin, type I Collagen, TGF-β1 and Type 4 collagen.
Values are expressed as (means ± SEM).
ADX, adriamycin.
*P < 0.01 vs. control; †P < 0.001; ‡P < 0.01 vs. ADX.

DISCUSSION
In the present study, we first demonstrated that a highly selective A3AR antagonist reduced proteinuria and albuminuria, improved renal function, and recovered histomorphological changes that occurred during ADX-induced nephropathy. We concluded that this selective A3AR antagonist had a notable effect on urinary nephrin excretion and its glomerular expression, which may prevent podocyte injury. Clinically, proteinuria is the most characteristic manifestation of podocyte injury in kidney disease. We used ADX to develop the renal glomerular injury. ADX induces proteinuria and nephrin loss in urine by injuring the glomerular filtration barrier, decreasing glomerular endothelial cell thickness and glomerular charge selectivity and increasing podocyte foot process effacement (1718). The renal histological changes due to ADX appear about one week after ADX administration and degenerate into severe injury by four weeks (19).
Adenosine has various effects in the kidneys, including the regulation of renin secretion, glomerular filtration rate, renal vascular tone, tubular glomerular feedback, fluid and electrolyte transport of the tubular and collecting duct system, and metabolic renal function. The concentration of extracellular adenosine is maintained at approximately 100 nM under normal conditions and rapidly increases during renal cell damage (20). A previous study demonstrated that long-term exposure to high concentrations of adenosine induced secretion of intracellular proinflammatory cytokines, activation of fibroblasts, and deposition of collagen in the extracellular matrix (21). A recent study showed that an increase in adenosine levels led to ischemic renal fibrosis and finally induced renal interstitial fibrosis in renal cell damage in the unilateral ureteral obstruction model (22).
Activation of A3AR by adenosine leads to a decrease in adenylyl cyclase activity and cAMP production, which induce stimulation of phospholipase C (PLC) activity, Ca2+ mobilization, and phosphorylation of phosphatidylinositol 3-kinase (PI3K) and AKT. AKT modulates the mitogen-activated protein kinase (MAPK) family including c-JUN N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 MAPK (6). Therefore, A3AR activation stimulates p38 MAPK phosphorylation, leading to decreased hypoxic damage of cardiomyocytes (23). On the other hand, A3AR activation suppresses the p38 MAPK pathway, leading to down-regulation of the inflammatory status in human synoviocytes (24). A3AR has different effects in different cell types, such as cell proliferation and cell death, which may be related to PI3K/AKT and ERK1/2 pathway crosstalk (25). Many studies have shown the biologic functions and therapeutic applications of A3AR and A3AR antagonists in the brain, heart, lung, and immune system (6).
LJ1888 dramatically suppressed the urinary excretion of nephrin. In immunohistochemical staining, the numbers of nephrin-positive cells were dramatically decreased by ADX and significantly restored by LJ1888 treatment. Nephrin is one of the molecules that constitute the podocyte slit diaphragm and it is involved the maturation of podocytes during glomerular development (26). Nephrin induces phosphorylation and activation of the PI3K-dependent AKT downstream signaling pathway. Several AKT targets bind to the phosphorylation-dependent 14-3-3 proteins, which are involved in the control of cell growth, migration, proliferation, and apoptosis. Nephrin-induced PI3K/AKT signaling plays a preventative role in podocyte apoptosis (27). A3AR transmits a cellular proliferation signal through the PI3K/AKT signaling pathway. Stimulated AKT is able to phosphorylate many downstream substrates such as Raf. Therefore, by blocking Raf through AKT activation and inducing cell death, A3AR inhibits the MAPK pathway, including RAS, Raf, MEK1/2, and ERK1/2 (25). A3AR gene expression in renal cortical tissue was dramatically increased in ADX-induced nephropathy. Although LJ1888 did not suppress A3AR gene expression, LJ1888 inhibited A3AR-associated PI3K/AKT and RAS/RAF/MEK/ERK downstream pathways, protecting podocytes from apoptosis. Additional studies are required to demonstrate the interaction of A3AR and nephrin downstream pathways.
There is a considerable body of evidence regarding oxidative stress in various kidney diseases. In ADX-induced nephropathy, oxidative stress and inflammation are important mechanisms of progressive proteinuric nephropathy leading to renal fibrosis (13). Renal tissue hypoxia induces the elevation of adenosine levels, leading to renal fibrosis (22). Oxidative stress induces stimulation of TGF-β1 expression and production of extracellular matrix, leading to glomerular and tubulointerstitial fibrosis. The MAPK family signaling is activated by oxidative stress in renal cells. The JNK pathway is associated with progression of renal fibrosis and tubular apoptosis (28). Stimulation of the ERK pathway is related to proliferative glomerulonephritis (29). Activation of the p38 MAPK pathway by IL-1, TNF-α, and TFG-β1 induces extracellular matrix accumulation, leading to tubulointerstitial fibrosis (30). A previous study demonstrated that LJ1888 significantly suppressed JNK and ERK phosphorylation during TGF-β1 upregulation in the kidney (9). In this study, we observed that LJ1888 treatment attenuated the increased urinary excretion of 8-isoprostane and the increased LPO concentration in kidney tissue after ADX injection.
In addition, mRNA expression of proinflammatory molecules including TLR4, TNFα, IL-1β, and IFN-γ increased in ADX-induced nephropathy, and dramatically recovered during LJ1888 treatment. The TLR4/NK-κB pathway is considered to be an important link between inflammation and oxidative stress in kidney disease (3132). Gene expression of profibrotic molecules including type IV collagen, PAI-1, MCP-1, TGF-β1, and NOX4, as wells as NF-κB protein, were increased by ADX and significantly decreased by LJ1888. Renal histological and immunohistochemical staining demonstrated similar results consistently. Type I collagen, type IV collagen, TGF-β1, and α-SMA accumulation in the fibrotic area of the renal glomeruli were increased in ADX-induced nephropathy, as was macrophage infiltration. Those profibrotic and proinflammatory molecules were genetically suppressed significantly by LJ1888 administration. The extensive glomerular and tubulointerstitial damage, including glomerulosclerosis, excessive collagen deposition, interstitial fibrosis, and tubular atrophy, were dramatically increased in ADX-induced nephropathy. In contrast, LJ1888 administration significantly ameliorated severe damage lesions in both the glomerular and tubular compartments. We suggest that LJ 1888 ameliorated oxidative stress, inflammation, and fibrosis in ADX-induced nephropathy through antioxidative mechanisms via A3AR.
In summary, we are the first to show that LJ1888, a highly selective A3AR antagonist, exhibits renoprotective effects by ameliorating podocyte injury, oxidative stress, inflammation, glomerulosclerosis, and tubulointerstitial fibrosis in ADX-induced nephropathy. Treatment with LJ1888 induced renoprotective effects by attenuating podocyte injury through decreased urinary excretion of nephrin. Therefore, we suggest that LJ1888 could be a new therapeutic drug for proteinuric renal disease. We recommend further study of LJ1888 in progressive renal disease to demonstrate its therapeutic effect more clearly.
 XML Download
XML Download