

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Wilson's disease is a rare autosomal recessive inherited disorder of copper metabolism causing excessive copper accumulation in the liver, brain, cornea and several other organs in the body, requiring specific treatment to remove or detoxify tissue copper and to prevent reaccumulation (1). Most patients with Wilson's disease present with acute or chronic liver conditions, while some present with major extrahepatic manifestations such as neurological disturbances, hematologic abnormalities, and psychiatric disorders. Others may show relatively rare conditions such as renal diseases, arthritis, cardiomyopathy, pancreatitis, and endocrine manifestations including hypoparathyroidism, abnormal menstruation, infertility, and hypoglycemia (12). Hypopituitarism has been reported sporadically in Wilson's disease, but cases we found clinically presented only as hypogonadotrophic hypogonadism or menstrual abnormalities. Here we report a case of a woman diagnosed with Wilson's disease initially presenting with psychiatric symptoms, also accompanied by hypopituitarism in the form of hypothyroidism and adrenal insufficiency.
CASE DESCRIPTION
A 40-year-old woman visited the department of psychiatry at our hospital on March 17, 2014 with depressive mood, general weakness, and loss of appetite. Her mother had been diagnosed with liver cirrhosis. She had received treatment for depression at a local clinic for 3 years. Her symptoms worsened over the past year as she refused to talk or eat.
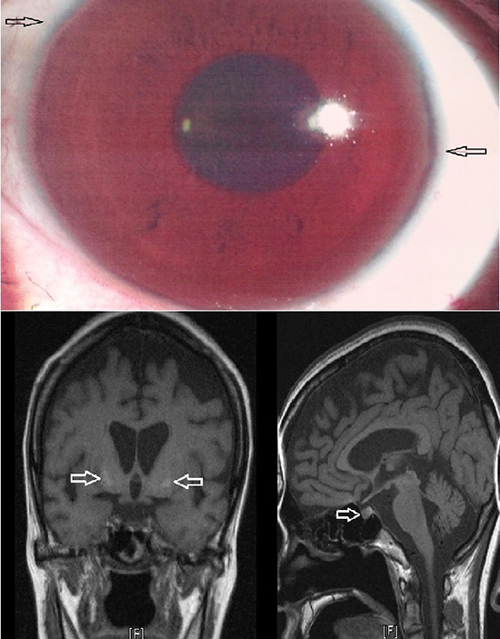
At admission, vital signs were stable and laboratory tests revealed the following results: hemoglobin 9.9 g/dL, hematocrit 30.5%, white blood cells 2,180/mm3, platelets 115,000/µL, total protein 5.4 g/dL, albumin 3.1 g/dL, BUN/creatinine 10.5/0.6 mg/dL, total bilirubin 1.1 mg/dL, direct bilirubin 0.47 mg/dL, AST/ALT 91/29 IU/L, prothrombin time 13.3 seconds (INR 1.22). An abdominal ultrasound was performed for further evaluation of low hemoglobin, thrombocytopenia and abnormal liver function test results, which showed liver cirrhosis and splenomegaly. As the patient had no history of alcohol consumption or recent use of hepatotoxic drugs, additional blood and urine tests were done to distinguish the cause of liver cirrhosis. The hepatitis B surface antigen, antibody, hepatitis C antibody, HIV and VDRL were all negative. Serum antinuclear antibody, Anti-smooth muscle antibody, LKM-1 antibody and anti-mitochondrial antibody were all negative and IgG was 824 mg/dL being in the normal range. Serum copper was decreased to 28.88 µg/dL (normal range: 90-130 µg/dL) and serum ceruloplasmin to less than 8 mg/dL (normal range: 20-40 mg/dL). The 24-hour urinary copper excretion was increased to 78.21 µg/day (normal range: < 40 µg/day). Slit-lamp examination revealed a Kayser-Flesicher ring, a band of golden pigment around the cornea which led to the final diagnosis of Wilson's disease (Fig. 1).

Basal hormone levels were checked in order to exclude other causes of depression, appetite loss, and general weakness. LH, FSH, estradiol, testosterone, IGF-1, and prolactin were all within normal range while thyroid function test results showed TSH 0.28 μIU/mL, fT4 0.72 ng/dL, and T3 50.85 ng/dL, all being below the lower limit. ACTH and cortisol were checked at 8 AM and 4 PM, being decreased to 1.00/1.72 μg/dL and 1.00/2.84 μg/dL, respectively. The thyrotropin releasing hormone (TRH) stimulation test and insulin tolerance test were performed to evaluate the pituitary-adrenal axis function. TSH levels showed only a marginal increase from 0.227 μIU/mL to 2.816 μIU/mL which was an insignificant response to TRH (Table 1), indicating central hypothyroidism and not non-thyroidal illness syndrome. ACTH remained as 1.00 pg/mL after provoking hypoglycemia with insulin injection while cortisol merely rose from 1.77 µg/mL to 1.66, and GH from 1.39 mg/dL to 9.78 mg/dL (Table 2). This was determined as insufficient response to insulin-induced hypoglycemia. Results were consistent with secondary hypothyroidism and adrenal insufficiency due to hypopituitarism.
Table 1
Result of TRH stimulation test (TRH 400 μg IV)

Table 2
Result of insulin tolerance test and baseline hormonal profile (regular insulin 0.1 U/kg IV)
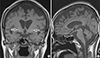
After being admitted, the patient showed trouble swallowing food and other neurological symptoms that could not be explained by depression such as rigidity, tremor and bradykinesia. Brain MRI was done as an additional test for these symptoms as well as to determine the presence of any pituitary tumors. T2-weighted images showed high signal intensity in both basal ganglia and in the midbrain, whereas no abnormality was found in the pituitary (Fig. 2).
Fig. 2
Bilateral symmetric increased signals of the basal ganglia on T2 weighted MR image (A) while no focal lesion was found in the pituitary gland (B).
Oral D-penicillamine (1.0 g/day) was given for copper chelation with pyridoxine (50 mg/day), while thyroid hormone and steroid replacement with levothyroxine (50 μg/day) and prednisolone (7.5 mg/day) were additionally used. Significant improvements were seen in liver function. Increase in 24-hour urinary copper excretion was monitored which reached 922.8 μg/day after initiating treatment and was maintained in the range of 200–500 μg/day after 6 months. Anti-Parkinson agents were used for tremor and gait disturbance. The patient's general weakness, appetite loss and depressive mood improved as well. She is currently receiving treatment with the same regimen and has had no side effects from medication or other newly developed symptoms for the last 7 months.
DISCUSSION
Hypopituitarism refers to decreased secretion of pituitary hormones as a result of abnormalities in the pituitary itself or the hypothalamus (3). It usually occurs due to a pituitary tumor or as a consequence of its treatment, and symptoms vary depending on which hormone is deficient (3). Other causes include pituitary apoplexy, Sheehan's syndrome, stroke, traumatic brain injury, cerebral hemorrhage and infiltrative diseases such as sarcoidosis or hemochromatosis which are relatively rare and are often wrongfully labeled as idiopathic since imaging is usually normal (34). In our case, hormone studies were consistent with secondary adrenal insufficiency and hypothyroidism due to hypopituitarism which we concluded to be associated with Wilson's disease, since no other definite cause or coexisting disease was found. The patient had no headaches, visual field defects or disturbances and brain MRI was negative of any pituitary lesion.
Among infiltrative diseases, hemochromatosis is known to cause pituitary dysfunction by depositing iron in the anterior pituitary, especially as hypogonadotrophic hypogonadism probably due to the preference for iron of gonadotrophic cells (3). As for our case, we suggest copper deposition or secondary neuronal damage in the pituitary to explain the association between hypopituitarism and Wilson's disease, but the mechanism could not be proven in this report. Recent studies showed that Wilson's disease may be complicated by high iron concentrations in the brain as low ceruloplasmin levels and reduced ferroxidase activity lead to iron overload along with copper in excessive amounts, causing oxidative damage in a synergistic cascade (5). Paris et al. (6) proposed dopamine-dependent neurotoxicity as another possible mechanism, decreasing survival of dopaminergic cells in the brain and contributing to the regional differences in copper accumulation or its consequent neuronal damage. In another report, a biomolecular basis of copper toxicity was presented, demonstrating that both copper and iron induce genome damage and inhibit its repair process (7). Whether these suggestions could be applied to the pituitary in Wilson's disease requires further investigation.
T2 hyperintense signals in the basal ganglia is the most common finding on the brain MRI in Wilson's disease, but interpretation of this lesion is ambivalent since copper deposition may be undetectable without the summation of underlying iron accumulation. Secondary neuronal damage might occur without direct local copper accumulation (8). Also, it is not uncommon for pituitary imaging to be normal in hypopituitarism (4). Despite normal pituitary imaging in our case, we could not rule out the possibility of copper infiltration as the cause for hypopituitarism in Wilson's disease.
Endocrinologic abnormalities are rare features in Wilson's disease. Symptoms such as delayed puberty or amenorrhea were sporadically reported, as well as glucose intolerance and parathyroid function disorders (910). Krysiak et al. (1) reported galactorrhea and menstrual abnormalities in a patient with Wilson's disease, presuming that the symptoms may have been caused by local deposits of copper in the pituitary and mammary glands. Frydman et al. (10) studied 16 patients with Wilson's disease to detect potential endocrine dysfunctions. In their report, most patients had low or borderline LH levels and a dynamic GnRH test revealed blunted LH and FSH responses. Also regarding a possible association between pituitary dysfunction and Wilson's disease, a case of delayed puberty in an 18-year-old male with Wilson's disease was reported, suggesting copper accumulation as the potential cause of decreased synthesis or secretion of GnRH in the hypothalamus or pituitary. Like in our study, the brain MRI was negative of any pituitary lesion (11). These cases presented pituitary dysfunction as hypogonadotrophic hypogonadism or menstrual abnormalities. To our knowledge, our study is an unprecedented case of Wilson's disease coexisting with hypopituitarism presenting as TSH or ACTH deficiency in Korea, as other pituitary hormones were maintained within the normal range.
Meanwhile, common psychiatric symptoms in Wilson's disease are depression, incongruous behavior, and cognitive impairment (12). It may be challenging to differentiate these symptoms from hypothyroidism or adrenal insufficiency which can be easily misdiagnosed or neglected if not examined for other potential conditions (13). This study highlights the importance of screening for hormone deficiency in Wilson's disease especially in patients presenting with psychiatric symptoms. We also suggest considering Wilson's disease when evaluating possible causes for idiopathic hypopituitarism. Regardless of whether the two conditions are causal or coincidental, both require treatment as soon as practicable.
Further confirmation is needed on the endpoint of hormone therapy and on whether or not copper-chelating treatment alone brings pituitary function recovery, as pituitary dysfunction was successfully reversed by liver transplantation or therapeutic phlebotomy in some cases of hemochromatosis. Also, it would be worthwhile to reevaluate the pituitary function in 6 to 12 months after discontinuing hormone replacement. In conclusion, this case indicates a possible link between Wilson's disease and hypopituitarism presenting as altered thyroid homeostasis and pituitary-adrenal axis dysfunction.
 XML Download
XML Download