

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Traumatic brain injury (TBI) is a complex process that includes primary, secondary or additional injury, and regeneration (1). Secondary injury mechanisms include complex biochemical and physiological processes that are initiated by the primary insult and manifest over a period of hours to days (2, 3). The initial disturbances caused by TBI lead to apoptotic cell death and brain edema, but the molecular mechanisms underlying these processes are unclear. Brain edema is a pathological condition of increased water content resulting from various coexisting brain injuries, including ischemia, trauma, tumors, and infection. Aquaporins (AQPs) are transmembrane proteins that selectively allow the passage of water through the plasma membrane of fluid-transporting cell types throughout the body. Recent studies have suggested activation of MAPKs as a molecular mechanism for brain edema and neural cell death in both in vitro and in vivo traumatic injury models (4,5,6,7,8). MAPKs also participate in osmotic shock-induced stimulation of AQP4 and 9 in rat cortical astrocytes (8, 9).
Moreover, nuclear factor-κB (NF-κB) regulates transcriptional activity in the nucleus and is involved in various cellular activities. NF-κB triggers several anti-apoptotic genes that interrupt the apoptotic cascade at multiple levels (10, 11). Therefore, NF-κB nuclear translocation plays a pivotal role in the transcriptional regulation of cell survival and death. Agmatine is an endogenous polyamine formed by the decarboxylation of L-arginine by arginine decarboxylase (ADC). Agmatine is an endogenous clonidine-displacing substance, an agonist for both α2-adrenergic and imidazoline receptors, and selectively blocks N-methyl-D-aspartate (NMDA) receptors (12,13,14). Agmatine treatment was reported to have various biological actions and neuroprotective effects on multiple central nervous system (CNS) diseases (12, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25). We have previously reported that agmatine attenuates brain edema by limiting blood-brain barrier (BBB) disruption and blocking the accumulation of brain water content by lessening AQP1 expression after cerebral ischemia (22). In addition, agmatine facilitated the activation and nuclear translocation of NF-κB in astrocytes following oxygen/glucose deprivation (21). All of these cellular mechanisms may contribute to the neuroprotective effects of agmatine.
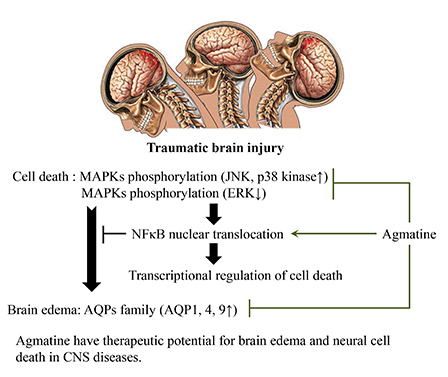
Based on these observations, we hypothesized that agmatine may have neuroprotective effects on brain edema and apoptotic cell death by suppressing AQP expression, MAPK phosphorylation, and promoting the translocation of NF-κB into the nucleus after TBI.
MATERIALS AND METHODS
TBI model
Eight-week-old male Sprague-Dawley rats (290±20 g, SAMTAKO, Osan, Korea) were subjected to TBI. Experimental animals were anesthetized with a mixture of Zoletil 50 (24 mg/kg) and Rompun (16 mg/kg). Anesthetized rats were placed on a conventional stereotactic frame, and a craniotomy was performed over the right forelimb motor cortex. Craniotomy was carried out by drilling +2 mm anterior and -2 mm posterior from Bregma and +1-5 mm lateral from the midline (Fig. 1). The bone was thinned with a grinder bit over the forelimb motor cortex, and the dura mater was left intact to prevent bleeding. TBI was induced by placement of a metal probe (3 mm in diameter) cooled by liquid nitrogen onto the right forelimb motor cortex. The motor cortex was exposed to the cooled probe five times for 30 sec of injury and 30 sec of rest (26). The physiological parameters of the animals were monitored and maintained before injury. All of the experimental animals were maintained under a 12-hr light/dark cycle and were sacrificed for analysis 1, 2, or 7 days after injury.
Agmatine administration
Agmatine (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in normal saline and injected intraperitoneally (100 mg/kg) 30 min after TBI and once daily until the end of the experiment. The daily dosage and timing of agmatine administration were determined from previous experiments (22, 25). Experimental control animals received normal saline in equivalent volumes (Fig. 1).
Determination of BBB disruption
Animals were injected intravenously with Evans Blue solution (2% in saline; 4 mg/kg; Sigma-Aldrich) at the onset of reperfusion and incubated for 60 min under anesthesia. Animals were perfused transcardially with normal saline. Coronally dissected 2 mm thick brain sections were analyzed using a computer-assisted image analysis system (Image J 1.36b; NIH, Rockville Pike, MD, USA).
Measurement of swelling volume and brain water content
Experimental animals were sacrificed to analyze maximal brain edema 1 or 2 days after TBI (18, 21, 22). Coronally dissected 2 mm thick brain sections were examined for the swelling volume and brain water content. The swelling volume (%) was calculated as follows: (volume of ipsilateral hemisphere/volume of contralateral hemisphere)×100.
To determine the brain water content, whole brains were separated sagittally and weighed to obtain the wet weights (WWs) and dry weights (DWs) of each hemisphere before and after drying at 110℃ for 24 hr, and the water content (%) was calculated as follows: (WWs-DWs)/WWs×100 (27).
Measurement of apoptosis by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining
TUNEL staining was performed following the manufacturer's instructions (In Situ Cell Death Detection Kit; Roche Diagnostics, Indianapolis, IN, USA). Briefly, brain sections were deparaffinized, rehydrated, and washed with phosphate buffered saline (PBS). Proteinase K was then added, and sections were incubated for 30 min at 37℃. Sections were washed twice with PBS, stained with the TUNEL reaction mixture for 60 min at 37℃, and washed twice with PBS. DNA fragmentation was observed under a fluorescence microscope (LSM 510 META; Carl Zeiss, Oberkochen, Germany).
Behavior tests
To examine the change in motor function, all of the animals were tested after TBI using the rotarod and limb placement tests. Animals were assessed on an accelerating rat-sized rotarod (ENV-577; Med Associates, Inc., St. Albans, VT, USA). The rod speed was gradually accelerated from 4 to 40 rpm. The limb placement test has been described elsewhere (28). Briefly, forelimb and hindlimb placement was evaluated under seven different conditions. A total of 0 points indicated maximal neurological deficit, and 24 points indicated normal performance. Animals were tested five times on each testing day with a minimum of 5 min of rest in the home cage between trials.
Observation of protein expression levels by immunohistochemical staining
Brains were fixed with a 4% paraformaldehyde solution, embedded in paraffin, and sectioned (10 µm). Sections were immunostained with primary antibody to either p-ERK (#9101), p-JNK (#9255), or p-P38 (#4631; all from Cell Signaling, Danvers, MA, USA) followed by an appropriate biotinylated secondary antibody. Brain sections were visualized using the ABC kit (Vector, Burlingame, CA, USA) and reacted with diaminobenzidine substrate (DAB; Sigma-Aldrich). To determine the fluorescent immunohistochemistry, brain sections were immunostained with primary antibodies (AQP1 [AB9566; Abcam, Cambridge, UK], AQP4 [AB3068; Millipore, Bedford, MA, USA], AQP 9 [AB3091; Millipore]) and visualized using a fluorescence-conjugated secondary antibody. Nuclei were counterstained with 4', 6-diamidino-2-phenylindole (DAPI). Sections were visualized using a fluorescence microscope (LSM 510 META; Carl Zeiss, Oberkochen, Germany).
Measurement of protein expression by immunoblotting
Brain tissues were dissected from the animals, homogenized, and lysed with ice-cold RIPA buffer and protein inhibitors (2 µg/mL aprotinin, 5 µg/mL leupeptin, 1 µg/mL pepstatin A, 1 mM phenylmethanesulfonyl fluoride [PMSF], 5 mM EDTA, 1 mM EGTA, 5 mM NaF, 1 mM Na3VO4), and incubated on ice for 60 min. Protein concentrations in the supernatants were determined using bicinchoninic acid (BCA) protein assay kits (Thermo Scientific, Rockford, IL, USA), and equal amounts of protein were subjected to electrophoresis on 10%-12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels. Separated proteins were electro-transferred to polyvinylidene difluoride membranes (Millipore), and then the membranes were blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS). The membranes were incubated overnight with a specific primary antibody against ERK (#9102), p-ERK (#9101), JNK (#9252), p-JNK (#9255), p-P38 (#4631; all from Cell Signaling), NF-κB p-65 (AB1606; Millipore), or β-actin (AB8226; Abcam) as indicated. Immunoreactive bands were detected with horseradish peroxidase (HRP)-conjugated secondary antibodies and visualized using the West Pico chemiluminescent substrate kit (Thermo Scientific). The band intensities were quantified using Image J software (Image J 1.36b; NIH, Rockville Pike, MD, USA).
Measurement of cytosolic/nuclear fractionized protein expression by immunoblotting
Brain tissues were dissected from the animals, homogenized, and lysed with ice-cold lysis buffer A (pH 7.9; 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT], 0.05% NP40 and protein inhibitors [2 µg/mL aprotinin, 5 µg/mL leupeptin, 1 µg/mL pepstatin A, 1 mM PMSF, 5 mM EDTA, 1 mM EGTA, 5 mM NaF, 1 mM Na3VO4], and then were incubated on ice for 15 min. After incubation, the lysates were centrifuged at 14,000×g for 20 min at 4℃. Supernatants were collected for the cytosolic fractions, and pellets were resuspended in lysis buffer B (pH 7.9; 5 mM HEPES, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 26% glycerol [v/v], 300 mM NaCl and protein inhibitors [2 µg/mL aprotinin, 5 µg/mL leupeptin, 1 µg/mL pepstatin A, 1 mM PMSF, 5 mM EDTA, 1 mM EGTA, 5 mM NaF, 1 mM Na3VO4]) and incubated on ice for 30 min. After incubation, the lysates were then centrifuged at 24,000×g for 20 min at 4℃, and the supernatants were collected for the nuclear fractions. The protein concentrations in the lysates were determined using BCA protein assay kits (Thermo Scientific), and samples containing equal amounts of protein (40 µg) were subjected to electrophoresis on 10% SDS-PAGE gels. Separated proteins were then electro-transferred to Immobilon-NC membranes (Millipore), and the membranes were next blocked with 5% BSA in TBS. The membranes were incubated overnight with a specific primary antibody against NF-κB p65 (AB1606), β-actin (AB8226), or histone H1 (sc-8030) as indicated. Immunoreactive bands were detected using HRP-conjugated secondary antibodies and visualized using the West Pico chemiluminescent substrate kit (Thermo Scientific). The band intensities were quantified using Image J software (Image J 1.36b; NIH, Rockville Pike, MD, USA).
Statistical analysis
Data were presented as the mean±standard error of the mean (SEM) of at least three different experiments, and duplicate or triplicate determinations were performed in each experiment. Statistical tests were performed with Student's t-test using SAS ver. 8.01 (SAS Institute Inc., NC, USA). A P value less than 0.05 was deemed to indicate statistical significance.
Ethics statement
All of the animal procedures were carried out according to a protocol approved by the institutional animal care and use committee (IACUC) of Yonsei University Animal Research Center (YLARC) using National Institutes of Health (NIH) guidelines. All of the animals were maintained in a specific pathogen-free facility of the YLARC (IACUC Approval No. 2011-0212).
RESULTS
Effect of agmatine on BBB disruption, brain edema, and apoptotic cell death after TBI
To determine whether agmatine treatment attenuates BBB disruption and brain edema after TBI, Evans Blue extravasation and brain water content were analyzed. Agmatine treatment reduced the area of Evans Blue extravasation (39.27 mm2; 17.9 %) 7 days after TBI (Fig. 2A and B), and the swelling volume was significantly decreased (5.24 mm2; 5.2%) 7 days after TBI (Fig. 2C) compared with that of control animals. The brain water content was reduced in the agmatine treatment group at 1 and 2 days after TBI (Fig. 2D). Agmatine treatment significantly reduced the necrosis area (3.56±0.1% for 2 days, 2.46±0.2% for 7 days; Fig. 2E and F) and the number of TUNEL-positive cells at 2 and 7 days after TBI (Fig. 2G) compared with that of the control animals.
Effect of agmatine on neurological motor function after TBI
To investigate whether agmatine treatment reduces the deficits in motor functional behavior following TBI, rotarod and limb placing tests were performed. Compared with control animals, animals treated with agmatine showed a significant behavioral improvement from 1 to 7 days after TBI (Fig. 3A) as measured by the rotarod test. However, the results of limb placement test for the evaluation of motor coordination were not significantly different in agmatine-treated compared with control animals from 1 to 7 days after TBI (Fig. 3B).
Effect of agmatine on the expression of AQPs after TBI
To investigate whether agmatine attenuates brain edema formation after TBI by regulating AQPs, the expression of AQP1, 4, and 9 was analyzed. We found that AQPs were significantly decreased in the agmatine treatment group 7 days after TBI using immunohistochemistry (Fig. 4A-C). In addition, the protein expression of AQPs was suppressed in the agmatine treatment group at 7 days after TBI as measured by immunoblotting (Fig. 4D).
Effect of agmatine on the phosphorylation of MAPKs after TBI
To investigate whether agmatine regulates the phosphorylation of MAPK after TBI, the expression of MAPKs was analyzed by immunohistochemistry and immunoblotting. Agmatine treatment suppressed the number of JNK- and P38 MAPK-positive cells at 1, 2, and 7 days after injury but activated the phosphorylation of ERK expression (Fig. 5A-C). By contrast, pJNK and pP38 MAPK expression was significantly suppressed in the agmatine treatment group from 1 to 7 days after TBI (Fig. 5D-G).
Effect of agmatine on nuclear translocation of NF-κB after TBI
To investigate whether agmatine regulates the nuclear translocation of the NF-κB p65 subunit after TBI, the expression of NF-κB p65 was analyzed by immunoblotting. Agmatine significantly decreased the cytosolic expression of NF-κB p65 but increased the translocation of NF-κB p65 to the nucleus 1 day after TBI (Fig. 6A-D).
DISCUSSION
In the present study, we hypothesized that agmatine may have neuroprotective effects on apoptotic cell death and brain edema after TBI. Previous studies have suggested that agmatine treatment decreases the size of infarcts in a mouse ischemia model and promotes survival in neurons after oxygen-glucose deprivation (OGD) (18), reduces excitotoxicity in vitro by blocking NMDA receptor activation (15, 19), and protects neurons from injury (12) by the inhibition of nitric oxide synthase (18). The present study used TUNEL staining to determine agmatine treatment decreased TUNEL-positive cells 2 to 7 days after TBI. It is possible that cells undergoing necrotic cell death could also be improperly labeled by TUNEL staining, although this result suggests that agmatine ameliorated neuronal cell death after TBI. In addition, recent studies have implicated the MAPK pathway in the progression of acute brain damage after ischemia or trauma. The expression of MAPK proteins was activated by various stress stimuli, including bacterial LPS and osmotic shock (6, 8). Generally, ERK is centered on multiple signal transduction pathways to accomplish a variety of functions, including proliferation, differentiation, and survival. Activation of JNK and p38 kinase signaling pathways tends to promote apoptosis, whereas activation of ERK signaling pathway tends to be anti-apoptotic role in the nucleus (29, 30). In this study, agmatine treatment suppressed the phosphorylation of JNK and p38 kinase, whereas activation of phosphorylated ERK after TBI. These results suggests that agmatine could regulates transcription of apoptosis and necrosis by MAPKs after TBI. The next question arises concerning how agmatine regulates transcriptional activation and apoptosis by suppressing MAPK phosphorylation after TBI. NF-κB translocation was investigated to analyze transcriptional activation and anti-apoptotic gene regulation. Previously, NF-κB was shown to trigger several anti-apoptotic genes that interrupt the apoptotic cascade at multiple levels (10, 11) and to play a pivotal role in the regulation of cell survival and cell death. In addition, strong evidence supports that NF-κB functions as an anti-apoptotic transcription factor in various cell populations, including neurons and astrocytes (10, 31, 32). Our results suggest that agmatine treatment has an anti-apoptotic effect by activating NF-κB nuclear translocation in TBI. Nuclear translocation of NF-κB should lead to suppress the apoptotic cell death after TBI.
Furthermore, accumulating evidence has shown that TBI could induce the breakdown of the BBB, a pathological hallmark of neurotrauma from in vivo and in vitro studies (33, 34). TBI is generally considered to the vasogenic origin of edema, sequential secondary injury promotes the opening of the BBB (35). Our previous study suggested that agmatine attenuated brain edema by limiting BBB disruption and blocking the accumulation of brain water content by decreasing AQP1 expression after cerebral ischemia (22). In addition, AQP4 is primarily expressed in astrocytes at the BBB, where it plays a role in water transport between fluid compartments within the brain (36). AQP4-null mice are partially protected from brain edema caused by hyponatremia or ischemia (37). Thus, dysfunction of AQP4 protects against cytotoxic (cellular) brain edema with intact BBB (38). AQP9 is also found on astrocyte processes and has specific permeability potential to lactate and water. AQP9 could contribute to lactic acidosis because it is permeable to water and lactic acid under pathological conditions (38, 39). Therefore, AQPs control water movement and are a potential therapeutic target for brain edema. Recent report suggested that MAPK pathways mediated cellular osmolarity by regulating aquaporin family. ERK activation is necessary for AQP5 induction by hyperosmotic stress (8). Phosphorylated P38 MAPK is also a key regulator of AQP4 and AQP9 under hyperosmotic and ischemic conditions in cultured cortical astrocytes (8, 9). Based on these observations, our results suggest that agmatine attenuated brain edema via AQPs through MAPK pathways under a pathological condition. In conclusion, we suggest that agmatine has neuroprotective effects by attenuating necrosis, BBB disruption, and brain edema after TBI. These neuroprotective effects involve reducing the expression of phosphorylated MAPKs and AQP1, 4, and 9 while promoting nuclear translocation of NF-κB after TBI. Therefore, agmatine treatment may have beneficial effects following various CNS injuries.





 XML Download
XML Download