

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Traumatic brain injury (TBI) is a major cause of death and persistent disability, and functional recovery often requires extensive rehabilitation (1). Although direct mechanical insult is the main mechanism behind TBI, additional programmed cell death or apoptosis may lead to neuronal and glial cell loss (23). Previous trials demonstrated that anti-apoptotic effects improved histological and functional outcomes after TBI (45). In addition, neuronal plasticity has a major role in functional recovery after TBI (67).
Repetitive transcranial magnetic stimulation (rTMS) is a non-invasive intervention that modulates neural connectivity. Rapid magnetic field changes in the stimulation coil induce electric currents in cortical areas, which are thought to reversibly modulate neuronal circuits by depolarizing cortical neurons (8910). Previous clinical trials showed that rTMS has beneficial effects on functional recovery after stroke (111213). One of the proposed therapeutic mechanisms of rTMS is anti-apoptosis, putatively by increasing B-cell lymphoma 2 (Bcl-2) expression (1415).
Till now, there are few trials which have examined the effects of rTMS on motor recovery after TBI. The purposes of this study were to investigate the effect of rTMS on behavioral recovery during the early stage of TBI in a rat model.
MATERIALS AND METHODS
Adult male Sprague-Dawley rats (n=20, 240-280 g) were housed in laboratory cages in a controlled environment (22.0-24.0℃) and were maintained under a 12/12 hr light/dark cycle with free access to food and water.
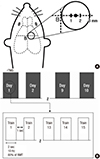
Rats underwent lateral fluid percussion for induction of TBI as described previously (16). Briefly, rats were anesthetized with an intraperitoneal injection of 1.0% ketamine (30 mg/kg, Ketara; YuhanYanghang, Seoul, Korea). A heating pad was used to maintain the body temperature at 37±0.5℃ throughout the procedure. The skull was cleaned with a topical depilatory agent and betadine solution, and a 3-mm craniectomy was performed 1.5 mm to the right of the midline bregma over the primary motor cortex without penetrating the dura (Fig. 1A) (17). A female Luer-Lock fitting was secured over the craniectomy site with ethyl methacrylate. A saline-filled injury device (Model 211 B4, Kistler Instrumental Corp., Amherst, NY, USA) was attached to the rat via a Leur-Lock. A single moderate severity pulse (3.5-4.0 atm pressure) was delivered by rapidly injecting a bolus of saline into the craniectomy site.
Twenty TBI rats were randomly assigned to either the rTMS (n=10) group or the sham (n=10) group. The rTMS group received 10 sessions of stimulation over a 2-week period beginning on the fourth day after TBI. Each daily session consisted of 15 trains of 2 sec at a rate of 10 Hz with a 1-sec inter-train interval (a total of 3,000 impulses; Fig. 1B). The stimulation coil was set tangentially above the stimulation site. For sham stimulation, the TBI rats were stimulated with a sham coil that produced the same noise as the real magnetic coil.
Rotarod and beam balance tests were performed on post-injury days 4 (before rTMS), 18 (after ten sessions of rTMS), and 25 by a blinded investigator. Brain magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) were performed on post-injury days 4, 18, and 25. There were no fatalities in either group during the experiment. Rats were sacrificed after the last evaluation, and the brains were collected for western blot and immunohistochemical analyses.
A Magstim rapid2 magnetic stimulator (Magstim Co., Whitland, Defeld, UK) with a figure-of-eight coil (12-mm inner diameter, 20-mm outer diameter, 3.5-T peak magnetic field) was used in conscious rats, as described previously (15). Biphasic stimulation with a clockwise current was applied using a 400-µs pulse. Motor evoked potentials (MEPs) in the biceps femoris muscle of the weak hind limb were measured using a Sapphire Premier device (Medelec, Old Woking, Surrey, UK), by a previous described method (18). Briefly, the rats were stimulated at the site at which the stimulus of a slightly suprathreshold intensity produced the largest peak-to-peak MEP amplitude in the biceps femoris.
The effects of rTMS on cortical modulation also depend on the stimulation intensity. Previous trials in which 80% to 130% of the resting motor threshold (RMT) was applied showed therapeutic benefits in stroke management (19). To maximize the safety of rTMS for treatment of TBI, the stimulation intensity used in this study was 80% of the RMT, the lowest possible therapeutic intensity. Additionally, our previous study showed that 80% of the RMT intensity had beneficial effects on motor recovery in a stroke rat model (15). RMT was defined as the lowest stimulator output at which the peak-to-peak MEP amplitude was greater than 5% of its maximal amplitude in at least five of the ten trials.
Rotarod and beam balance tests were performed to assess motor coordination and balance ability. Rats were put on the rotarod at an initial speed of 4 rpm, and the speed was increased to 40 rpm over the course of four minutes. For the beam balance test, rats were placed in the center of a wooden rectangular beam (60 cm in length and 1.5 cm in diameter), which was set horizontally and suspended 40 cm above a foam pad. In both tests, the time to fall was measured. Each test was administered three times by a blinded researcher, and the results were averaged.
Rats were placed on a 4.7-T Bruker Biospec Imager (Bruker Medical Systems, Karlsruhe, Germany) and anesthetized with an intraperitoneal injection of 1.0% ketamine. Intact hemispheric volume (VIH), lesioned hemispheric volume (VLH), and lesion volume were measured using Paravision 3.0 software (Bruker Biospec, Ettlingen, Germany) by a blinded rater. Tissue-processing artifacts were minimized by calculating the edema ratio as follows: edema ratio=VLH/VIH×100 (20). The voxel of interest was selected over the peri-lesional site (3×3×3 µL) of the subcortical area on a T2-weighted image (T2WI). All raw spectroscopic data were processed using the Bruker's XWIN-NMR software installed in the MRI system. The metabolites were identified by the chemical shifts as observed in the MRS. The values in parts per million were as follows: choline (Cho), 3.2 ppm; creatine (Cr), 3.03 ppm; and N-acetyl-aspartate (NAA), 2.0 ppm. Cho and NAA peak areas were quantitated as relative ratios to Cr, which is constant in the brain tissue and used as a standard (21).
At post-injury day 25, rats were anesthetized with intraperitoneal injection of 1.0% ketamine and perfused transcardially with 50 mL of phosphate-buffered saline (PBS) and another 50 mL of 4% paraformaldehyde in 0.1 M/L PBS. The brains were removed, fixed in 10% neutral-buffered formalin, dehydrated in a graded ethanol series, and embedded in paraffin for immunohistochemistry, and they were obtained from the peri-lesional area. Each block was sectioned at a 6 µm-thickness and mounted on poly-L-lysine coated slides. These slides underwent a serial process of deparaffinization with xylene, dehydration with ethanol, washing with saline, and microwave in citrate buffer for 10 min. Hydrogen peroxide was used to block endogenous peroxidase activity for 30 min to reduce the non-specific immunoreactivity. Primary antibodies against B-cell lymphoma 2 (Bcl-2, 1/100, Abcam, Cambridge, MA, USA) and Bcl-2 associated X protein (BAX, 1/100, Abcam) were employed. Bcl-2 and BAX are anti-apoptotic proteins, as previously described (22). Anti-mouse, HRP/DAB (Labvision, Fremont, CA, USA) was used as the secondary antibody. Immunostained sections were digitized using a 400×objective (Leica DFC 290, Leica, Heerbrugg, Germany) in conjunction with the Leica Application suite (version 3.3.0, Leica). Five views obtained from the peri-lesional area were evaluated by a blinded pathologist, and the stained cells were counted and averaged.
Brains were removed at post-injury day 25 and frozen at -20℃ for western blots. The tissues from the peri-lesional area in brains were selected for the blots. Proteins were extracted with a series of procedures including mixing with the protein extraction solution, homogenization by vortexing for 5 min, and harvesting the supernatants after centrifugation for 10 min at 15,000 rpm. Total protein amounts were assayed with the Bradford's method. Proteins were resolved on a SDS-PAGE gel, and blotting was performed for 90 min. Primary antibodies were applied onto the nitrocellulose membrane for 2 hr. After washing, secondary antibodies were added to the membrane for 1 hr. Primary antibodies against B-cell lymphoma 2 (Bcl-2, 1/100, Abcam) and Bcl-2 associated X protein (BAX, 1/1,000, Abcam) were applied for protein analyses. An antibody for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Relative band density was assessed with Image J software.
Statistical analysis was performed with PASW software package, version 18.0. Student's t-test was applied to evaluate the differences in behavioral tests, edema ratio, and metabolites between the two groups on post-injury day 4. Repeated-measures ANOVA was used to analyze behavioral tests, lesion volume, edema ratio, and relative ratios of metabolites. Immunostaining and western blot data were analyzed using the Student's t-test. P values less than 0.05 were considered significant.
RESULTS
Behavioral tests
Baseline rotarod and beam balance data on post-injury day 4 did not show significant differences between the rTMS and sham groups. There were no significant group×time interactions for the rotarod and beam balance tests among two groups (Fig. 2).
There were no significant differences in baseline MRS data for Cho/Cr and NAA/Cr ratios between the two groups. Group×time interactions for Cho/Cr and NAA/Cr ratios were not significant among two groups (Fig. 3).
Brain MRI
Baseline data for brain swelling on post-injury day 4 did not show significant differences between the two groups. There was no significant group×time interaction for lesion volume or brain swelling between the groups (Fig. 4).
Immunohistochemistry
Immunohistochemical analyses of Bcl-2 and Bax staining showed that the rTMS group had a significantly greater number of Bcl-2 stained cells and a lesser number of BAX stained cells around the peri-lesional area compared with the sham group (P<0.05; Student's t-test; Fig. 5).
Western blot
The densitometric analyses of western blots showed that the rTMS group had a significantly higher Bcl-2 expression and a lower BAX expression compared with the sham group (P<0.05; Student's t-test; Fig. 6).
DISCUSSION
This is the first report to investigate whether application of rTMS for TBI has a therapeutic effect on motor recovery or not. The major finding is that rTMS did not improve motor function during the early stage of TBI, although an anti-apoptotic effect was observed in the peri-lesional area without aggravation of brain injury.
Previous experiments showed that rTMS has an anti-apoptosis in the peri-ischemic areas in models of acute and subacute ischemic stroke (1415). The peri-ischemic region, known as 'ischemic penumbra', is functionally silent due to impaired blood supply, but it sustains minimal metabolic activities (23). Therefore, most of the apoptosis is observed in the ischemic penumbra (242526). In this study, the rTMS group demonstrated significantly enhanced expression of Bcl-2 and reduced expression of BAX, which are known the anti-apoptotic and pro-apoptotic proteins, respectively (Fig. 5) (222728). Apoptosis triggered by TBI leads to progressive neuronal loss (23). However, the observed anti-apoptosis did not induce motor recovery in rat TBI model, which could indicate that the mechanical damages causing necrosis of tissue, cell swelling, and plasma membrane rupture were more significant than apoptosis in early stage of TBI (29). These could be more serious when the intensity of trauma is severe. Therefore, anti-apoptosis induced by rTMS might be unable to suppress direct cellular injury during the early stage of moderate TBI. The present results are in agreement with those of previous reports, in which Bcl-2 over-expression did not elicit functional improvement in a TBI model (303132).
Additionally, the damaged brain might be more vulnerable to excitation during the early injury period. Previous studies showed that early exercise aggravated brain injury in an animal model of stroke (3334). Considering the previous trials which reported that TBI patients require a longer recovery time than stroke patients (3536), it can be argued that the application of rTMS to the damaged brain during the early period of injury counteracted the beneficial effects of anti-apoptosis induced by rTMS after TBI.
Cho and NAA measured by 1H MR spectroscopy are the structural component of cellular membrane and a predecessor of brain lipids, respectively (37). In states of neuronal damage, including TBI, the Cho level is expected to increase with a decrease of the NAA level. In this study, Cho/Cr and NAA/Cr ratio measured by MRS were not significantly different among the groups (Fig. 3). Additionally, MRI findings showed that lesion volume and brain edema was not improved nor aggravated after rTMS (Fig. 4). These results meant that rTMS was neutral effect on neuronal recovery or deterioration in early stage of moderate TBI.
There are some limitations to this study. First of all, we did not include a healthy group. The inclusion of these groups could have explained the therapeutic effects of rTMS compared to those in the normal control group, and it could also have helped to clarify the effect of TBI on motor function. Additionally, adverse events such as seizures or tremors were not measured during the experimental period. Therefore, we could not report any other adverse events associated with rTMS, except for lesion volume, brain edema and metabolic changes.
In conclusions, rTMS did not have beneficial effects on motor recovery during early stages of moderate TBI, although an anti-apoptosis was observed in the peri-lesional area. Although behavioral data did not support the claim that rTMS is beneficial for treatment of TBI, further studies should be performed in different periods or severities of TBI, using diverse rTMS protocols and more sensitive behavioral tests to evaluate the potential therapeutic effects of rTMS.




 XML Download
XML Download