

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth frequently diagnosed malignancy and is the second leading cause of cancer death worldwide. Several environmental factors, such as hepatitis B and C viral infections, exposure to aflatoxin B1, and alcohol abuse, are major causes of HCC, but standardized therapeutics have not been established for HCC, except surgery and liver transplantation (1). A wide variety of genetic or epigenetic alterations and aberrant regulation of oncogenes or tumor suppressor genes are associated with the multistep progression of HCC, but the molecular pathogenesis of HCC remains poorly understood (23).
Histone modifications, such as acetylation, methylation, and phosphorylation of lysine residues, play critical roles regulating gene expression. Among these modifications, acetylation of histones is a major epigenetic alteration involved in transcriptional regulation. Acetylation of histones is balanced by the action of two enzyme families, such as histone acetyltransferases (HATs) and histone deacetylases (HDACs). HDACs, which remove acetyl groups from histone lysine residues, are implicated in transcriptional repression of a wide variety of genes (45). HDACs consist of four classes, such as class I (HDAC1, 2, 3, and 8), class II (HDAC4, 5, 6, 7, 9, and 10), class III (SIRT1-7 and sirtuins), and class IV (HDAC11), based on the homology of yeast histone deacetylases (4). HDACs induce the formation of heterochromatin without DNA binding by associating with other transcription factors. Interestingly, sirtuins deacetylate many non-histone proteins, such as p53, MyoD, FOXO3, NF-κB, and others (6). Because of their broad range of substrates, HDACs are implicated in the initiation and progression of human diseases including cancers. Aberrant regulation of oncogenic or tumor suppressive HDACs has been suggested in various human cancers, such as gastric cancer, lung cancer, and HCC (78910). Thus, understanding the regulatory mechanisms of HDACs in cancers is important for developing therapeutic agents to treat these cancers.
MicroRNAs (miRNAs) are small regulatory non-coding RNAs of 21-25 nucleotides. MiRNAs are generated from their precursor transcripts via a series of processing steps. Mature miRNAs mediate mRNA degradation or suppress mRNA translation by binding to the 3'-untranslated region (3'-UTR) of target mRNAs (11). Due to their wide variety of target genes, miRNAs affect many biological pathways, including cell proliferation, development, and differentiation. Deregulation of miRNAs facilitates cancer development by upregulating oncogenes or silencing tumor suppressor genes (12). Aberrant expression of miRNAs, such as miR-21, miR-29, and miR-221, regulates tumor cell growth, apoptosis, migration, and invasion in HCC by targeting proteins involved in those cellular pathways (131415). Furthermore, emerging evidence suggests that miRNAs inhibit expression of HDACs by directly targeting HDAC transcripts in human cancers.
In this review, we provide additional insights into the roles of HDACs, particularly in HCC. In addition, we describe the regulatory mechanisms of HDACs, focusing on post-transcriptional regulation by miRNAs and discuss the therapeutic potential of HDACs and HDAC-regulating miRNAs in liver malignancies.
HDACS IN HUMAN CANCERS
Aberrant HDAC expression in human cancers
Epigenetic regulators, such as HATs, HDACs and sirtuins, histone methyltransferases, histone demethylases, histone variants, and chromatin remodeling factors, have arisen as major regulators of gene expression in cancer research. The expression or activity of epigenetic regulators is disrupted in human cancers. In particular, three HDACs (HDAC1, HDAC2, and HDAC6) and four sirtuins (SIRT1, SIRT2, SIRT3, and SIRT7) are closely implicated in human tumors among HDACs (16).
Aberrant expression of HDACs generally disrupts cellular biosystems and occasionally initiates cancer and progression. That is, global rearrangements of DNA methylation and histone modifications are representative marker of cancer development. HATs and HDACs are the most frequent deregulated molecules among epigenetic modulators, as they are responsible for chromatin dynamics by adding or removing acetyl groups from lysine residues in the amino terminal tails. Hyperacetylated histones are generally located in active genes and hypoacetylated histones are found in silent regions of heterochromatin. Acetyl modification of histones reflects different cell status and, in some cases, the alterations may precede the cell transformation by changing the expression of tumor suppressors and oncogenes leading to initiation of cancer (16).
Aberrant expression of HDACs is correlated with cancer aggressiveness and poor prognosis (17). Many studies have suggested aberrant expression of HDACs in diverse cancers. Upregulation of HDACs is significantly correlated with key events during cancer onset and poor disease-free and overall survival. For example, functional loss of adenomatosis polyposis coli (APC), a tumor suppressor, increases HDAC2 expression and leads to tumor transformation, such as a lack of apoptosis in colorectal cancer (18). The HDAC2 protein level is overexpressed in lung cancer tissues and is associated with oncogenic properties, such as tumor cell growth and activation of cellular apoptosis (819). Similarly, HDAC2 is upregulated in gastric cancer tissues and targeted inactivation of HDAC2 reduces tumorigenesis by restoring p16INK4a activity (920). Moreover, our previous study reported that HDAC2 is negatively correlated with p21WAF1/Cip1 regulation and that aberrant expression of HDAC2 disturbs homeostasis by dysregulating gene expression of cell cycle components in liver cancer (10)
Some HDACs function as oncogenes or tumor suppressors, depending on the cellular context and tumor type. For example, SIRT3, a mitochondrial NAD-dependent deacetylase, is significantly overexpressed in oral squamous cell carcinoma cells and human tumor tissues, and inhibiting SIRT3 represses cell growth and proliferation in vitro and in vivo (21). In contrast to the oncogenic role of SIRT3 in oral cancer, SIRT3 transcript levels are decreased in breast cancer, and ectopic expression of SIRT3 suppresses cell growth by regulating hypoxia inducible factor target genes, supporting a tumor suppressive role for SIRT3 (22).
Although many studies have reported that abnormal expression and mutation of HDACs are closely related to tumorigenesis in many cancers, more effort is needed to delineate their biological roles and molecular mechanisms during cancer development.
Pivotal roles of individual HDACs in liver cancer
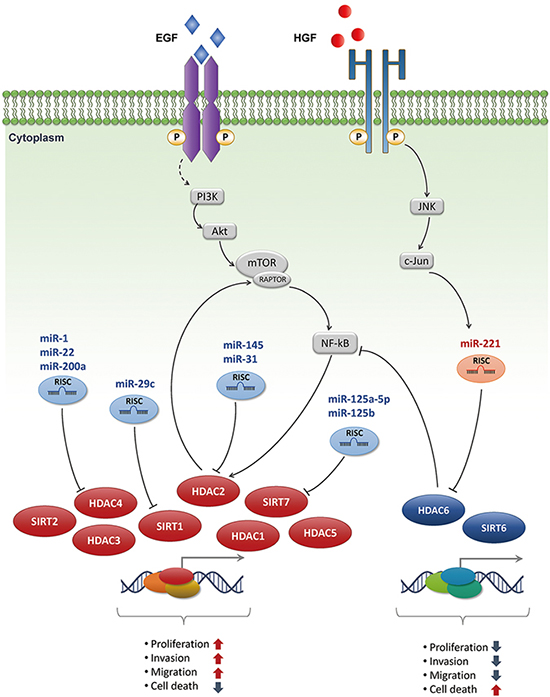
Emerging evidence indicates that expression of HDACs is remarkably disrupted in many human cancers. Lachenmayer et al. reported that expression of a subset of HDACs is increased in liver cancer compared to normal liver tissues with the presence of cirrhotic and dysplastic nodules (23). In addition, HDAC3 and HDAC5 DNA copy numbers are altered and their expression levels are significantly upregulated in HCC (24). Concomitantly, our group has described diverse roles of HDACs in HCC for several decades. In a comprehensive gene expression profile analysis of clinical samples of multi-step hepatocarcinogenesis, HDAC1, HDAC2, and SIRT7 were upregulated from pre-neoplastic lesions to high-grade HCCs, whereas HDAC6 was gradually downregulated (Fig. 1) (Table 1) (25).
In the previous study, HDAC8, a member of Class I HDACs, is overexpressed in liver cancer. HDAC8 knockdown repressed tumor cell growth and induced apoptosis through p53 expression and acetylation at Lys373 (26). Another study revealed that HDAC5 mRNA and protein levels are overexpressed in human HCC tissues and that inhibition of HDAC5 represses growth of HCC cell lines. Suppression of HDAC5 induces apoptotic cell death and G1/S cell cycle arrest by regulating apoptosis-associated molecules and cell cycle regulators (27). Similarly, depletion of HDAC2 selectively induces p16INK4a and p21WAF1/Cip1, leading to inhibited G1/S cell cycle transition. Furthermore, knockdown of HDAC2 causes hypophosphorylation of pRb and, consequently, E2F/DP1 target genes, such as CDC2, PCNA, and E2F1, are downregulated (10). mTROC1/NF-kBp50 signaling is related to growth factor-induced HDAC2 expression and is sustained in HCC. mTORC1 activity is maintained by HDAC2 stabilizing the mTOR/RAPTOR complex, and AKT (serine/threonine-specific protein kinase) is phosphorylated through a positive feedback loop triggered by HDAC2 (28). HDAC1 is overexpressed in a subset of human HCCs and liver cancer cell lines. Inactivating HDAC1 results in regressed tumor cell growth and activation of caspase-independent autophagic cell death, through the LC3B-II activation pathway in Hep3B cells (29). In this regard, overexpression of HDAC1 and HDAC2, which are class I HDACs, may play a pivotal role by regulating mitotic effectors during development of HCC.
SIRT7 is a class III HDAC, which requires the NAD+ cofactor for catalytic activity. SIRT7 was initially known as a nuclear protein associated with ribosomal gene transcription (30). SIRT7 expression was upregulated gradually in a large cohort of patients with HCC (25). Furthermore, depletion of SIRT7 inhibits in vitro and in vivo HCC tumorigenesis by selectively regulating the cell cycle and autophagic molecules in HCC cells. That is, SIRT7 may control tumor progression or development of cancer phenotype (31). SIRT2, another member of the sirtuin family, is also upregulated in HCC cell lines and in a subset of human HCCs. Overexpression of SIRT2 in HCC tissues is significantly associated with the presence of microscopic vascular invasion, an advanced tumor stage, and poor survival rates (32). Unlike the oncogenic functions of SIRT7 and SIRT2 in HCC, decreased SIRT6 expression is observed in primary human liver cancers and apoptosis-insensitive hepatoma cell lines. Ectopic expression of SIRT6 in the HepG2 liver cancer cell line increases apoptotic cell death. Moreover, loss of SIRT6 induces global hypomethylation and metabolic changes (2633).
Although anomalous expression of individual HDACs, such as HDAC1, HDAC2, and HDAC6, have been reported in many cancers, the roles of HDAC6 in cancer development are still hold both oncogene and tumor suppressor. In our recent study, HDAC6 expression was downregulated in patients with HCC, and was significantly related with poor prognosis for 5-yr overall, disease-free, and recurrence-free survival. Intriguingly, ectopic overexpression of HDAC6 significantly suppressed cell growth in liver cancer cell lines and induced LC3B-II conversion and caspase-independent autophagic cell death through the JNK/Beclin1 pathway. This finding suggests that HDAC6 is involved in tumor suppression by nonepigenetic regulation in hepatocarcinogenesis (7).
Regulation of HDAC expression by miRNAs and therapeutic approaches
MiRNAs are a class of small noncoding RNAs that negatively regulate gene expression. Many miRNAs have emerged as key regulators associated with liver tumorigenesis. For example, Pineau and his group profiled the expression of miRNAs in 104 HCC tissue samples and 35 HCC cell lines (15). Of the upregulated miRNAs, miR-221 was the most upregulated miRNA in tumor samples. MiR-221 targeted the CDK inhibitor p27Kip1 and DNA damage-inducible transcript 4 (DDIT4), inhibiting cell proliferation when an anti-miR specific for miR-221 was treated to liver cancer cell lines. Similarly, a variety of miRNAs were identified as oncogenes or tumor suppressors governing aberrant expression of key epigenetic regulators in hepatocarcinogenesis (Fig. 1) (Table 2). For example, HDAC4 has critical roles in cancer development and has been identified as a direct target of miR-1, miR-22 and miR-200a in HCC (223435). Additionally, miR-31 and miR-145 are significantly downregulated in a subset of primary HCCs, and ectopic expression of these miRNAs exerts an anti-tumor effect by directly targeting oncogenic HDAC2 in vivo and in vitro (3637). SIRT1 is aberrantly regulated in HCC and its overexpression stimulates HCC cell growth via inactivating transcription of p21Cip1, p27Kip1, p15INK4b and activating CDK2, CDK6, cyclin D3, and cyclin D1. Notably, although the SIRT1 protein is highly overexpressed in patients with HCC, SIRT1 mRNA expression is not significantly different between HCC and non-tumor groups. Additional research evidenced that miR-29c was an endogenous regulator of SIRT1 and its expression was significantly downregulated in a large cohort of HCC patients (38). Up-regulation of SIRT7 exerts the tumorigenic effect of HCC and its expression is negatively correlated with miR-125a-5p and miR-125b, which directly regulate translation of SIRT7 by binding the 3'-UTR of its transcript (31). In addition to these tumor suppressive miRNAs targeting oncogenic HDACs, a recent study identified that repression of the tumor suppressor HDAC6 was directly regulated by miR-221 via coordinated JNK/c-Jun- and NF-κB-signaling pathways in the progression of liver tumorigenesis (8).
Many groups have suggested possible clinical applications for miRNA-based cancer therapy. Although a plethora of inhibitors have been developed and utilized to treat HCC, suitable drugs for liver cancer therapy are needed because overall survival rates of patients with liver cancer remain low. For example, sorafenib is the standard treatment for advanced HCC, but there are many side effects to be solved, such as subcorneal pustular dermatosis (39). Furthermore, cancerous cells occasionally circumvent drug targeted signaling by adapting to diverse environments, which is linked to poor patient prognosis (40). Accumulating evidence indicates that strategies based on modulating miRNA are a possible approach for liver cancer therapy. Elmén et al. showed simple systemic delivery of anti-miR and effectively antagonized liver-expressed miR-122, a tumor suppressive microRNA, in non-human primates without toxicity (41). This treatment resulted in prolonged survival and reduced the size and the number of tumors. Furthermore, systemic delivery of miRNA using adeno-associated virus vehicle is another attractive method for liver cancer therapy. If miRNAs could be delivered in viral vectors and continuously transcribed, they may sustain high expression of miRNA mimics or anti-sense miRNAs in target tissues. One study reported that miR-26a, which is downregulated in HCC, was delivered successfully to the liver and reduced the tumor burden in a c-Myc-induced murine liver cancer model (42). However, many additional miRNAs that successfully inhibit liver tumorigenesis remain to be characterized. That is, a single miRNA or a cluster of miRNAs governing a subset of oncogenes initiating or developing cancer may be a promising candidate for cancer therapeutic delivery. For example, miR-188 efficiently blocks the G1/S cell cycle transition by directly targeting multiple cyclins, such as cyclin D, cyclin E, and cyclin A, in nasopharyngeal cancer cells (43). Similarly, miR-7 remarkably suppresses the downstream epidermal growth factor receptor pathway by regulating multiple molecules, such as PI3K, phosphorylated AKT, Raf-1, and phosphorylated MEK 1/2, in glioma cells (44). Similarly, because of the critical roles of HDACs in cancer development, if miRNAs that dominantly regulate these effectors could be found and specifically delivered to a target tissue, they may have novel cancer therapeutic applications.
In this review, we summarized the role of the HDAC families and HDACs targeting miRNAs during the development and progression of liver cancer. Accumulating evidence suggests that HDACs regulate the expression and activities of a variety of proteins involved in cancer development. Furthermore, certain HDACs are anomalously expressed in tumor tissues and have redundant functions in liver cancer (232425). Despite the many drugs that inhibit HDAC activities, overall survival rates and prognoses of patients with HCC remain poor. That is, liver cancer cells easily acquire drug resistance because they adjust to their environment through transformation (4546). Thus, miRNAs could be an efficient therapeutic approach for liver malignancies. Although technological progression asserts that using miRNAs or anti-miR as therapeutics is practical and safe, more studies are needed to move the field toward clinical applications.


 XML Download
XML Download