

 PDF
PDF ePub
ePub Citation
Citation Print
Print



The functional integrity of the endothelium is a fundamental element of vascular health. Many studies have established that endothelial dysfunction (ED) is not only an initiator, but could also be an important factor in the progression of atherosclerotic cardiovascular disease (1). Generally, the risk for atherosclerotic cardiovascular disease is estimated on the basis of traditional cardiovascular risk factors (2). However, these risk factors are thought to account for about 50% of the pathogeneses of atherosclerotic cardiovascular diseases, which means unknown risk factors have a substantial role in atherogenesis (3). Thus, a risk assessment for cardiovascular disease that depends only on traditional risk factors may be insufficient to identify an individual's current risk. Much evidence has been reported that the progression of ED is related to the intensity and duration of the patient's risk factors, and to the amount of total risk in individual subjects (4). Moreover, ED independently predicts cardiovascular events in addition to traditional risk factors (56). In this respect, a patient-specific assessment of endothelial function could give us the actual current status of a patient's cardiovascular risk and progression of atherosclerosis.
ROLE OF THE ENDOTHELIUM
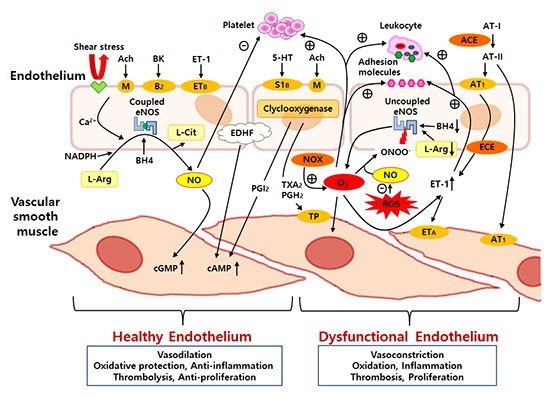
The vascular endothelium is an active monolayer of cells covering the lumen of blood vessels, separating the vascular wall from the circulating blood. For many years, the endothelium had been believed to be a simple cellular barrier. However, extensive research has revealed the endothelium's complex role. The endothelium is a highly selective barrier and metabolically active organ, and it plays a crucial role in the maintenance of vascular homeostasis by keeping a delicate balance between vasodilation and vasoconstriction (7). The vasodilaton is mainly mediated by factors such as nitric oxide (NO), endothelium-derived hyperpolarizing factor (EDHF), and prostacyclin, while a vasoconstrictory state is mediated by factors such as endothelin-1 (ET-1), angiotensin II, thromboxane A2, and prostaglandin H2 (8). An overview of the effect of vascular endothelial factors on the function of vascular smooth muscle and circulating blood cells is shown in Fig. 1. Among these endothelial-derived factors, NO is considered to be the most potent endogenous vasodilator in the body (9). In addition, NO also maintains the homeostasis of the vascular wall by inhibiting platelet aggregation, inflammation, oxidative stress, vascular smooth muscle cell migration and proliferation, and leukocyte adhesion (10). Basically, NO is produced and released from L-arginine through the activity of the endothelial NO synthase (eNOS) under the influence of chemical agonists acting on specific endothelial chemoreceptors or by mechanical forces on mechanoreceptors, such as shear stress (11).
Chronic exposure to cardiovascular risk factors and oxidative stress overwhelms the defense mechanisms of the vascular endothelium, which is followed by ED and the loss of the endothelium's integrity, smooth muscle cell proliferation and migration, and leukocyte adhesion and migration (12). Much evidence has been pointing to ED as one of the major pathologic changes between exposure to the cardiovascular risk factors and the development of atherosclerotic cardiovascular disease. In addition, ED is observed in the early stage of most cardiovascular diseases (13). Therefore, the endothelium is a front line organ and endothelial function shows us an integrated index of all atherogenic and atheroprotective factors present in an individual (6). Hence, early clinical detection of ED may become a critical point in the prevention of atherosclerosis and cardiovascular disease because early detection of ED could be an initial reversible step in the development of atherosclerosis (Fig. 2).
PROPOSED MECHANISMS OF ENDOTHELIAL DYSFUNCTION AND PROGRESSION TO AHTEROSCLEROSIS
Role of NO and oxidative stress
The progression from ED to atherosclerosis is complex and multifactorial. As the endothelial function deteriorates, vascular homeostasis becomes impaired and leads to reduced anti-oxidant and anti-inflammatory effects, increased vascular permeability to lipoproteins, and the increased expression of inflammatory cytokines and adhesion molecules (14). Among various complex mechanisms, oxidative stress appears to be the most common underlying mechanism for the development of ED. Usually, most cardiovascular risk factors are associated with the up-regulation of intracellular oxidative stress and reactive oxygen species (ROS), which promotes several mechanisms, such as the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, NO inactivation, formation of peroxynitrite (ONOO-), uncoupling of eNOS, stimulation of endothelin expression, and so on (15). In the healthy endothelium, the eNOS is responsible for most of the vascular NO production. However, eNOS becomes a potential ROS generator when in the pathological uncoupled state (Fig. 1).
Chronic inflammation
Inflammation is another common underlying mechanism of ED, and there seems to be a causal relationship between oxidative stress and inflammation (16). Under physiological conditions, the endothelium controls vascular inflammation by releasing NO. However, a dysfunctional endothelium will promote ROS generation and aggravate vascular inflammation, which is detrimental to the vascular system. Oxidative stress may amplify the vascular inflammation signaling pathways, and inflammatory cells increasingly release superoxide (16). There are numerous inflammatory markers associated with endothelial dysfunction and atherosclerosis. C-reactive protein (CRP) is a primitive acute phase inflammatory protein that is released in response to various kinds of inflammation. There was a report that CRP directly contributes to the early phase of atherosclerosis by deposition on the intima, which precedes the appearance of monocytes (17). In addition, CRP directly affects NO bioavailability, which causes oxidative stress, endothelial dysfunction, and intimal hyperplasia. A possible mechanism was proposed that CRP directly acts through lectin-like oxidized low-density lipoprotein (oxLDL) receptor-1 which plays a pivotal role in oxLDL-induced endothelial dysfunction in human aortic endothelial cells (18). Inflammation is also associated with the overexpression of tumor necrosis factor-alpha (TNF-α) and interleukin-1, which promotes leukocyte adherence and migration (19). In addition, these inflammatory cytokines induce endothelial cells and leukocytes to express adhesion molecules, such as vascular cell adhesion molecules (VCAM) and intercellular adhesion molecules (ICAM), monocyte chemotactic protein-1, E-selectin, P-selectin, and interleukin-6, resulting in a worsening of ED (20). After reaching the intima, monocytes transform into macrophages and express receptors that facilitate the uptake and accumulation of lipids, which lead to the transformation of the macrophages into foam cells. Later, smooth muscle cells migrate, and then fatty streaks formed by macrophage-derived foam cells and vascular smooth muscle cells, which are the precursor lesion of atheroma, may become atheromatous plaque (21). Thus, ED plays a key role in both the initiation and progression of atherosclerotic plaque.
Infection
Endothelial dysfunction and atherosclerosis are also affected by infection and its immune-mediated injury (22). Epidemiological studies indicate infectious agents may predispose patients to atherosclerosis and its clinical events. Viruses, such as cytomegalovirus and herpes simplex virus-1, and bacteria, such as Chlamydia pneumoniae and Helicobacter pylori, have reported to be associated with coronary artery disease (CAD) in humans (23). It has been shown that immunoglobulin-G antibody titers to cytomegalovirus, hepatitis A virus, herpes simplex virus-1, Chlamydia pneumoniae, and Helicobacter pylori were independent risk factors for endothelial dysfunction and the presence of CAD. Therefore, it is likely that during infection with these pathogens, there may be shared crucial immunologic pathways by which diverse organisms produce endothelial injury and arthrosclerosis (2425).
Vitamin D deficiency
Among several cardiovascular risk factors, vitamin D (1α, 25-dihydroxycholecalciferol) deficiency is emerging as a new candidate. Vitamin D appears to participate indirectly in atherosclerosis and systemic inflammation. Including endothelial cells, vitamin D receptors (VDRs) are present in all cells implicated in atherosclerosis (26). In patients with subclinical atherosis and slow coronary blood flow, a strong association was found between vitamin D deficiency and endothelial dysfunction (27). Molinari et al. reported that vitamin D was found to stimulate NO production in human umbilical venin endothelial cells through eNOS activation. This effect was the VDR-mediated phosphorylation of intracellular kinases, such as p38 and protein kinase B, leading to eNOS activation. Recently, in vivo and in vitro experiments have demonstrated that a vitamin D analog (22-oxacalcitriol) significantly suppressed the elevated expression of NADPH oxidase and improved eNOS coupling, thus reducing oxidative stress in the endothelium (28). Moreover, vitamin D protected endothelial cells against H2O2 oxidative stress, counteracting superoxide generation and apoptosis.
Shear stress
Although the entire vascular tree is exposed to the systemic risk factors of ED, atherosclerotic lesions usually generate at specific arterial regions, such as bifurcations, branching points, and the inner aspect of curved segments of the coronary artery (29). Locally disturbed shear stress by pulsatile blood flow is one of the modulators of the atherogenic process and accounts for the regional and clinical variability of atherosclerosis (30). Local endothelial shear stress (ESS) triggers vascular phenomena that synergistically exacerbate atherosclerosis toward an unstable phenotype (29). Specifically, low ESS modulates endothelial gene expressions through mechanoreception and mechanotransduction processes, inducing an atherogenic endothelial phenotype and the formation of early atherosclerotic plaque (31). Low ESS leads to atherosclerosis by augmenting ET-1 and suppressing NO, prostacyclin production, and lipid uptake and its catabolism, as well as induces plaque inflammation and oxidation in endothelial cells (32). As low ESS is primarily associated with plaque progression and vulnerability, higher ESS values are considered atheroprotective through the up-regulation of eNOS. Therefore, increases in vascular blood flow and shear rate are considered the main mechanisms of the beneficial effects on the cardiovascular system during exercise (33). However, excessively increased ESS also seems to be associated with plaque vulnerability. There have been reports that increased shear stress values have been associated with plaque ruptures or intimal ulcerations in the coronary and carotid arteries (34).
CARDIOVASCULAR RISK FACTORS AND ENDOTHELIAL DYSFUNCTION
Endothelial dysfunction has been reported in relation to most risk factors for atherosclerosis, such as diabetes, dyslipidemia, hypertension, smoking, aging, and obesity (835).
Diabetes mellitus
Generally, diabetes is an independent risk factor for the development of atherosclerosis and cardiovascular disease. In addition, it has been known that hyperglycemia impairs endothelial function (36). Interestingly, even in normoglycemic subjects who have a high risk for developing diabetes and insulin resistance, ED had been observed during an oral glucose tolerance test (37). In the patients with diabetes, the mechanisms of ED were a decreased synthesis of NO and increased production of vasoconstrictor substances (38). Guzik et al. reported that in patients with diabetes, oxidative stress, NADPH oxidase, and eNOS uncoupling had an important roles in developing ED (39). Hyperglycemia also leads to the generation of advanced glycation end products (AGEs), which are the products of the non-enzymatic glycation of proteins and lipids. AGEs accumulate in the vessel wall, alter the structural integrity of the endothelium and basement membrane, and are able to impede NO activity. This contributes substantially to ED. In addition, AGEs bind to specific surface receptors, which are expressed on cells such as monocytes, macrophages, and vascular smooth muscle cells, resulting in the amplification of an inflammatory response, increased vascular permeability, and oxidative stress (3640).
Dyslipidemia
High levels of LDL cholesterol and low levels of HDL cholesterol are independently associated with ED and inflammation (41). In patients with coronary artery disease and dyslipidemia, the increased degradation of NO by ROS has been suggested because the infusion of the L-arginine (substrate of NO) partially normalized the impaired coronary endothelial function (42). Possible mechanisms underlying dyslipidemia-induced ED include: (1) the up-regulation of NADPH oxidase, oxidative stress, and increased O2- production; (2) increased plasma levels of asymmetric dimethylarginine (ADMA); and (3) the oxidation of LDL. ADMA is an endogenous inhibitor of eNOS and competes with L-arginine for the same binding site on eNOS, which results in eNOS uncoupling. As a result, O2- production is increased and NO production is decreased (43).
Hypertension
In patients with hypertension, some findings have been shown to lead ED, such as the activation of the vasoconstriction process including the endothelin system and the activation of the inflammatory system (44). In addition, the increased production of ROS and decreased NO bioavailability have both been observed in patients with hypertension (45). Recently, inflammation has been reported as having a causative relationship with the development of hypertension, and it has been found that inhibiting inflammatory pathways can impede the process of developing hypertension and ED associated with atherosclerotic disease (46). This also suggests that anti-inflammatory approaches could provide methods for reducing ED associated with hypertension.
Smoking
Cigarette smoke contains many free radicals and delivers free radicals directly to the wall of the blood vessel. Besides being the supplier of free radicals, cigarette smoke facilitates the endogenous release of ROS through the activation of inflammatory cells (47). Therefore, smoking is associated with oxidative stress, inflammation, and the release of circulating factors that are associated with endothelial injury and ED (48). Furthermore, smoking has been reported to decrease HDL cholesterol levels, which is known to have anti-atherosclerotic properties and improve endothelial function (49).
Aging
Aging is an important determinant of cardiovascular risk in both men and women, and increasing age has been considered as one of the main factors that predisposes people to ED (50). The mechanism of ED during the aging process is accompanied by deterioration in balance, which is mainly characterized by a reduction of NO bioavailability and an increase in the production of cyclooxygenase-derived vasoconstrictor factors. It has been reported that there is a reduced expression and activity of eNOS as well as a decreased expression of NO and its activity in older animals (51). While endothelium-derived contracting factors, such as ET-1 and cyclooxygenase-derived prostanoids, and ROS production are increased. Plasma levels of ADMA are also known to rise with increased age (52).
Obesity
Patients with obesity have an increased risk of atherosclerosis (53). It is conceivable that simultaneous insults of several cardiovascular risk factors will multiply the injury of endothelial cells. Endothelial injury in obesity is characterized by impaired endothelium-dependent vasodilation, increased vasoconstrictor activity of ET-1, enhanced prostanoid-mediated vasoconstriction, and inflammatory activation (5455). In 397 patients with normal or mild coronary CAD, obesity was independently associated with coronary ED (56). The treatment of obesity often improves or even normalizes obesity associated insulin resistance or hypertension, and doing so also improves ED (57).
ASSESSMENT OF ENDOTHELIAL FUNCTON
Direct measurement of endothelial function
The direct measurement of endothelial function has been an important clinical tool in diagnosing and predicting the development of cardiovascular disease. For this reason, endothelium-dependent vasomotion has been widely used as clinical end-point for the assessment of endothelial function. Initial clinical studies for the evaluation of endothelial function were performed invasively in coronary circulation (58). This method involves the intracoronary infusion of endothelium-dependent vasodilatory substances (such as acetylcholine) with the measurement of changes in vessel diameter using quantitative coronary angiography (59). The endothelial function of the coronary microcirculation can be evaluated by measuring coronary blood flow with an intracoronary Doppler wire during coronary angiography with the intracoronary infusion of endothelial agonists (60). Recently, non-invasive methods for the assessment of endothelial function were developed. An ideal method for the direct measurement of endothelial function should be safe, non-invasive, cost-effective, repeatable, reproducible, and standardized between laboratories (12). Currently, flow-mediated dilation (FMD) of the brachial artery is the most widely used vascular test to assess endothelial function (61). The strengths and weaknesses of the current methods of assessing endothelial function, including FMD, low-flow mediated constriction, endothelial peripheral arterial tonometry, and venous occlusion plethysmography are described in Table 1.
Circulating markers of endothelial function
A number of laboratory markers have been identified and used as indicators of ED, such as E-selectin, ICAM-1, VCAM-1, interleukin-1, tumor necrosis factor-α, interferon-γ, monocyte chemoattractant protein-1, the von Willebrand factor, tissue plasminogen activator, plasminogen activator inhibitor-1, microalbuminuria, and tests of apoptosis (7). However, only some may have potential clinical use, because of their non-specific character, they allow insight into multiple facets of endothelial physiology and represent a complement in the assessment of endothelial function.
Recently, ADMA has emerged as a mediator, independent risk factor, and potentially promising marker for ED. ADMA is endogenously synthesized via the methylation of arginine residues, and it competitively inhibits eNOS, resulting in decreased NO production, which may induce eNOS uncoupling (62). Because increased ADMA levels have been documented in patients with dyslipidemia, hypertension, and atherosclerotic cardiovascular disease, they might be a useful measure of endothelial status and a potential marker for cardiovascular risk in clinical practice (63). However, European Society of Cardiology Working Group recommended that direct measurements of endothelial function remain a superior indicator and should not be replaced with plasma ADMA levels due to inconsistent prognostic data (64).
A reduction in endothelial-derived NO production or bioavailability represents a measurable parameter that is suggestive of the development of ED. Laboratory-based studies most often make use of indirect NO measurements to confirm ED, such as levels of nitrogen oxide or nitrite and nitrate, which are stable degradation products of NO (65). However, they are difficult to measure and may not always represent endothelial NO production (66).
Noninvasive imaging techniques for endothelial function
Advances in newer imaging technologies, such as cardiac magnetic resonance imaging (CMR), positron emission tomography (PET), and multi-detector computed tomography (MDCT), promise non-invasive means to assess vascular endothelial function and early atherosclerosis. Compared to invasive cardiac craterization, noninvasive imaging techniques may allow for the evaluation of healthy or low-risk populations with serial studies in the same patient over time.
The measurement of myocardial blood flow (MBF) using PET with sympathetic stress makes it possible for the evaluation of endothelial function. Although the spatial resolution of PET is limited and does not permit direct visualization of the coronary artery at the same time, the changes in MBF can be quantified with PET and used to infer coronary artery vasomotor responses (67). MDCT offers a much higher spatial resolution than PET and is widely available for the assessment of coronary status. Myocardial perfusion CT has been useful in assessing myocardial ischemia with pharmacological stress induced MBF changes (68). A recent study demonstrated a significant correlation between MDCT-measured changes in the epicardial coronary cross-sectional area and PET measured changes in MBF during a cold pressor test (69). However, both PET and MDCT expose subjects to the risks of ionizing radiation, which may be an important limitation of use in low-risk populations.
CMR is an appealing noninvasive imaging technique for quantifying coronary endothelial function because it is a tomographic technique offering high soft tissue contrast, excellent spatial resolution, and the ability to quantify blood velocity and flow without exposing subjects to ionizing radiation (70). In humans, CMR has been validated to measure absolute coronary arterial flow (71). Recently, CMR has also been used to document endothelial dependent coronary artery vasomotor responses to isometric handgrip exercises for coronary artery disease (72).
ENDOTHELIAL THERAPY: GENERAL CONSIDERATIONS AND THERAPEUTIC POTENTIAL
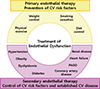
The treatment of ED is an approach to preserve or restore endothelial function. This concept allows us to interfere with the specific pathogenic pathways of the early time point of ED and to slow down the progression to atherosclerosis. It is important to note that although the underlying risk factors or severity of disease status may be different, these risk factors or disease conditions share several common pathophysiologic mechanisms that accelerate atherosclerosis (73). These factors are commonly associated with the activation of inflammatory pathways and reduced NO activity, mostly due to its inactivation by ROS (50). In addition, the activation and increased production of endothelium-derived vasoconstrictive factors such as ET-1 and angiotensin II, would change endothelial function toward a prothrombotic state (74). Thus, therapies for improving endothelial function will have beneficial effects on numerous cellular levels to impede the progression of atherosclerosis (175).
Endothelial therapy can be classified as (1) primary endothelial therapy for subjects without clinical cardiovascular disease, such as physical exercise, weight control, smoking cessation, and diet control, or (2) secondary endothelial therapy to improve dysfunctional endothelial homeostasis by treating and improving the underlying cardiovascular risk factors (control of hypertension, obesity, dyslipidemia, and diabetes) or established cardiovascular disease (treatment of atherosclerosis, heart failure, and renal failure), which are clearly supported by clinical evidence (Fig. 3).
Primary endothelial therapy: Modification of cardiovascular risk factors
Primary endothelial therapy could also be viewed as the preservation and improvement of endothelial function in subjects without cardiovascular risk factors. The major goal of this approach is to prevention of the cardiovascular risk factors with optimal lifestyle modifications, such as diet control, physical exercise, weight control, and smoking cessation.
Physical exercise
Recent studies have provided clear evidence that physical activity improves endothelial function, not only in healthy subjects but also in individuals at increased cardiovascular risk (76). Physical exercise also improves endothelial function and reduces cardiovascular risk in various conditions, such as older individuals, obese patients, postmenopausal women, and patients with dyslipidemia (19). In addition, exercise training leads to an improved bioavailability of the endothelium in patients with coronary artery disease and heart failure (77). In patients with severe congestive heart failure (n=40), a group that did four weeks of daily exercise training showed a significantly improved endothelial function compared to the control group (8.6% vs. 2.5%, P<0.001) (78).
Weight control
An altered endothelial cell phenotype and ED are common among patients with obesity (70). Short and long-term intervention studies have investigated the effects of weight control on endothelial function and have suggested that ED could be improved by successful medical and surgical weight loss. Biqomia et al. showed that in severely obese subjects who lost ≥10% body weight (BMI 48±9 kg/m2) demonstrated significant improvement in endothelial function in brachial FMD (from 6.8±4.2 to 10.0%±4.7%, P<0.01) (79). In 41 obese subjects treated with medical or gastric bypass operation, weight loss improved endothelial function and correlated strongly with lower levels of fasting glucose. The data demonstrate that weight loss in markedly obese patients improves endothelial function and glycemic control, which may reflect important mechanisms of the cardiovascular benefit with weight reduction (80).
Smoking cessation
Vascular dysfunction induced by smoking is initiated by reduced NO bioavailability, and further deteriorated by the increased expression of adhesion molecules (81). Much evidence for smoking-induced ED stem from clinical studies analyzing endothelial function. In 200 healthy young adults, Celermajer et al. reported that cigarette smoking was associated with the dose-related impairment of endothelial function (82). The reduction of the endothelium dependent dilatation from smoking was reversible. In 1504 current smokers, Johnson et al. showed prolonged improvement of endothelial function after smoking cessation of one year (83). In terms of secondhand smoke, passive smoking was shown to impair endothelial function in healthy young adults, which was shown to be reversible after the cessation of exposure (84).
Diet control
Diet appears to play an important role in the modulation of atherosclerosis and is associated with changes in endothelial function, which appear to be closely related to inflammatory processes (85). In the Nurse's Health Study, 732 participants enrolled to compare endothelial function in relation to two major dietary patterns. The prudent pattern was characterized by higher intakes of fruit, vegetables, fish, poultry, and whole grains, and the Western pattern was characterized by higher intakes of red and processed meats, sweets, desserts, french fries, and refined grains. Compared with the women who consumed the Western diet, the women who consumed the prudent diet were found to have significantly lower levels of C-reactive protein, ICAM-1, VCAM-1, and E-selectin, which are serum markers for inflammation and endothelial dysfunction. These findings are suggestive of a mechanism for the role of dietary patterns in the pathogenesis of ED and cardiovascular disease (86). Compared with Western diets, the Mediterranean diet comprised of fish, vegetables, fruits, and olive oil, has been found to reduce secondary cardiovascular events (87). In 180 patients with the metabolic syndrome, the Mediterranean diet was also found to have greater improvement in inflammatory markers and endothelial function when compared with a low-fat diet (88).
Secondary endothelial therapy: Control of cardiovascular disease progression
The goal of secondary endothelial therapy is to preserve the function of the already injured endothelium in order to delay the progression of atherosclerosis (1). Secondary endothelial therapy can be applied to both modifiable (hypertension, obesity, diabetes, dyslipidemia) and non-modifiable conditions (aging, menopause, coronary or peripheral artery disease, heart failure, end-stage renal disease).
Pharmacological treatment have reported effects on underlying disease conditions also show consistent results regarding restoring endothelial function, such as anti-hypertensive medication to control blood pressure, statin treatment to reduce LDL cholesterol, and antidiabetics to reduce blood glucose levels (59). It should be noted that some classes of pharmacological drugs can improve endothelial function, and in some instances reduce the risk of cardiovascular diseases. Modena et al. studied that in postmenopausal women with mild-to-moderate hypertension and impaired brachial FMD (n=400) who underwent 6 months of optimal control of blood pressure with antihypertensive therapy. After 6 months, FMD had not changed (≤10% relative to baseline) in 150 (38%) of 400 women (group A), whereas FMD had significantly improved (>10% relative to baseline) in the remaining 250 women (62%) (group B). During 67 months of follow-up, the patients in group A developed more cardiovascular events than patients in group B (3.50 per 100 person-years vs. 0.51 per 100 person-years, P<0.01) (89). As a secondary prevention setting, in 251 patients with newly diagnosed coronary artery disease with an impaired FMD of the brachial artery (<5.5%), 104 (41%) patients showed persistent impairment of FMD despite 6 months of optimized medical therapy. During 36 months of follow-up, cardiovascular events occurred in 27 (26%) patients with persistently impaired FMD and in 15 (10%) patients with improved FMD (P<0.01) (90). Thus, it is probably a good prognostic sign when ED is reversed with optimal treatments.
In patients with advanced atherosclerotic cardiovascular disease, percutaneous or surgical interventions should be applied to improve perfusion and shear-mediated endothelial function (91). Impairment of left ventricular function and attenuated peripheral perfusion is one of the important causes of endothelial dysfunction in patients with heart failure. A number of studies have reported that exercise training and optimal medical therapy improve not only exercise capacity and cardiac function but also endothelial function in patients with heart failure (9293). In patients with end-stage renal disease, dialysis should be considered to remove circulating toxic components to reduce endothelial injury (94).
Lipid-lowering therapy
The beneficial effects of statins on endothelial function have been extensively studied. In 1995, randomized trials demonstrated that lovastatin can restore the endothelial function of the coronary artery (95). The mechanism of statin-induced improvement of endothelial function seems to be independent of lipid-lowering effects. The advantageous statin effects on endothelial function are attributed to their anti-inflammatory and antioxidant properties (96). One study showed that simvastatin treatment resulted in a significant reduction in markers of endothelial dysfunction, inflammation, oxidative stress, and endothelial apoptosis. In this study, the reduction of CRP appears to be related to the lipid-lowering effects of simvastatin (97). It has been demonstrated that statins are able to increase the expression of eNOS and reduce ET-1 by inhibiting the expression of pre-pro ET-1 mRNA (98). Most of the statins have been shown to play an important role in the correction of ED (99). Early initiation of statin therapy can improve endothelial function, not only in patients with acute coronary syndrome, but also in patients with stable coronary artery disease (100101). A recent meta-analysis showed that statin therapy is associated with significant improvement in both peripheral and coronary endothelial function (102).
Angiotensin-converting enzyme inhibitor and angiotensin receptor blocker
Angiotensin-converting enzyme inhibitors (ACEIs) improve endothelial function by inhibiting the angiotensin-converting enzyme and reducing the production of angiotensin II (103). Studies on angiotensin receptor blockers (ARB) have demonstrated a positive effect on endothelial function, which supports the important role of angiotensin II in the development of atherosclerosis (104). In addition, ACEIs promote the stabilization of bradykinin, which induces the release of NO and prostacyclin, and reduces the production of oxygen free radicals through vascular NADPH oxidase, which is stimulated by angiotensin II (105). In the TREND study, compared to placebo, 6 months treatment of quinapril improved endothelial dysfunction in normotensive patients with coronary artery disease (106). These benefits are likely due to the attenuation of the vasoconstrictive effects and superoxide generating effects of angiotensin II and to the enhancement of endothelial cell release of NO secondary to the diminished breakdown of bradykinin. In the ISLAND study, ARB demonstrated the improvement of endothelial function and reduced inflammatory markers, which implicated an important role for it in the pathogenesis of atherosclerosis (104). A recent meta-analysis showed that ACEIs improve endothelial function in patients with ED caused by various conditions and are superior to calcium channel blockers and β-blockers. There was no significant difference between the effects of ACEIs and ARBs on peripheral endothelial function (107).
Renin inhibitor
In controlling the renin-angiotensin system, it remains unclear whether ACEIs and ARBs are fully effective. In this sense, renin inhibitors neutralize any compensatory increase in plasma renin activity and reductions in angiotensin I and angiotensin II levels (108). Aliskiren is a potent and specific inhibitor of human renin for the treatment of hypertension (109). There have been reports that aliskiren increases plasma NO bioavailability and endothelial function and suppress atherosclerotic plaque formation (110). Recently, a report had identified the effects of aliskiren on fructose-fed hypertensive rats. The results showed that aliskiren prevents and ameliorates insulin resistance, aortic endothelial dysfunction, and oxidative vascular remodeling in rat aortas (111).
Endothelin-I antagonist
Endothelin-1 is a 21-amino acid peptide with both mitogenic and vasoconstricting properties. ET-1 contributes to the regulation of vascular tone through two major receptors: endothelin-A (ET-A) and endothelin-B (ET-B). ET-A receptors are located on vascular smooth muscle and mediate vasoconstriction. ETB receptors are located on both endothelial cells, where they contribute to vasodilation through NO, and on smooth muscle cells, where they cause vasoconstriction (112). Predominantly, ET-1causes vasoconstriction via the activation of the ET-A receptor. There was a report that both systemic and coronary circulating ET-1 increased in patients with coronary endothelial dysfunction and early atherosclerosis (113). In addition, short-term and long-term blockade of ET-A receptor results in coronary vasodilatation and improvements in endothelial function (114).
Beta blockers
Unlike first- and second-generation β-blockers, third-generation β-blockers, such as carvedilol and nebivolol have favorable effects on endothelial function. Both drugs have been reported to stimulate β3 receptors, which activates eNOS and has antioxidant effects and the increased release of NO (115116). A recent randomized controlled study reported that compared with metoprolol, carvedilol significantly improved brachial endothelial function in patients with hypertension and type 2 diabetes mellitus when given for 5 months in addition to their current antihypertensive medications (117).
Estrogens
There have been reports about estrogens that are able to improve endothelial function through antioxidant properties that increase NO expression and promote the degradation of superoxide anion radicals (118). Estrogens are able to improve peripheral and coronary endothelial function, and this effect is independent from the beneficial effect on lipid profile, and they modulate vascular tone through a NO dependent mechanism (119). Sumino et al. reported that in osteoporotic postmenopausal women, the group that was treated with oral raloxifene for 12 months showed significantly increased brachial FMD compared with the control group (120).
In conclusion, the functional integrity of the endothelium is a fundamental element for vascular health. Many studies have established that ED could be an early integrated index of all atherogenic and atheroprotective indicators present in an individual. Until now, several biochemical markers and functional studies have led to the development of early detection and therapeutic interventions for ED. However, further research with large randomized controlled trials is necessary to aim at guiding the evaluation and treatment of ED. Ultimately, this effort could lead us to the prevention of atherosclerosis and changing of cardiovascular disease outcomes.

 XML Download
XML Download