

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Megalocytic interstitial nephritis is a rare form of chronic renal inflammatory disease associated with defect in intracellular destruction of invading foreign organisms by macrophages (1). These unusual inflammatory disorders are often associated with chronic urinary tract infection by Gram-negative bacteria (2). Although the pathogeneses of these diseases are unclear, macrophage bactericidal dysfunction has been presumed as a possible pathogenic mechanism (3). We report an extraordinary case of a 45-yr-old woman who had oliguric acute kidney injury (AKI) and acute pyelonephritis with Escherichia coli (E. coli) bacteremia, accompanied by megalocytic interstitial nephritis.
CASE DESCRIPTION
A 45-yr-old woman was hospitalized for abdominal pain, watery diarrhea, and jaundice of one-week duration on October 28, 2013. One day before admission, she noticed a marked reduction in her urine output. Her past medical history was unremarkable except for a two-year history of alcoholism.
At the time of admission, her blood pressure was 134/84 mmHg, and heart rate was 93 beats per minute. Her respiratory rate and body temperature were 20 breaths/min and 36.5℃, respectively. The patient was icteric and confused. She was unable to state her precise complaints. Physical examination was unremarkable except distended abdomen.
Laboratory findings included leukocytosis of 24,300/µL, high serum creatinine level of 6.80 mg/dL, high C-reactive protein level of 24.61 mg/dL, and high procalcitonin level of 39.05 ng/mL. Liver function test showed abnormalities including low albumin level of 2.9 g/dL, high aspartate aminotransferase level of 139 U/L, but alanine aminotransferase of 31 U/L. Coagulation time was prolonged to a PT INR of 1.46. Urinary findings showed hematuria and pyuria. Physical examinations and laboratory results suggested urosepsis with underlying alcoholic liver cirrhosis.
A computed tomographic (CT) scan of the abdomen revealed diffuse swelling of both kidneys with perinephric infiltration, suggestive of acute pyelonephritis. Additionally, hepatic nodularity and atrophic change as features of liver cirrhosis were observed.
The patient was diagnosed with acute pyelonephritis (APN) accompanied by oliguric AKI. Antibiotic treatment using cefotaxime (third-generation cephalosporin) and azithromycin was administered for a presumed diagnosis of APN, Weil's disease, and rickettsial infection. Continuous renal replacement therapy (CRRT) was initiated for septic shock with oliguric AKI. After recovering from septic shock, the patient was switched to conventional hemodialysis for the treatment of AKI. Later, E. coli susceptible to all feasible antibiotics were isolated from initial urine and blood cultures. Azithromycin administration was discontinued because there was no evidence of elevation of tsutsugamushi and leptospira antibodies.
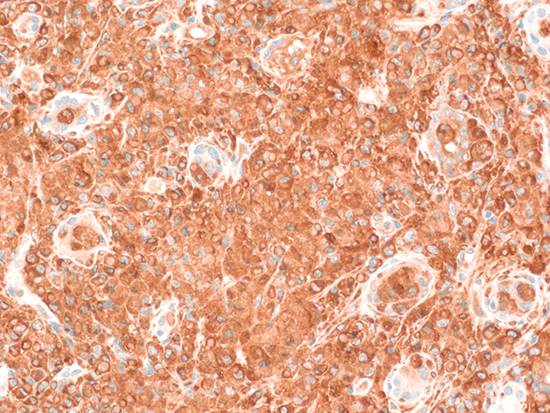
Although there was no microorganism in subsequent blood and urine cultures with improving parameters of infection such as fever, CRP, and procalcitonin, there were persistent severe leukocytosis with atypical lymphocytes and high level of lactate dehydrogenase over 1,000 IU/L. Serum protein electrophoresis (PEP) revealed increased gamma-globulin (34.5%), and serum immunofixation (IF) showed an abnormal band against anti-immunoglobulin G (IgG) and anti-lambda. To evaluate hepatosplenomegaly and blood cell abnormalities including leukocytosis, thrombocytopenia, and monoclonal gammopathy (<3 g/dL), bone marrow examination was performed. Bone marrow examination showed hemophagocytic histiocytes and increased plasma cells (below 10%). These results suggested the possibility of monoclonal gammopathy of undetermined significance because there was no evidence of bone lesion and hypercalcemia. After one month, kidney biopsy was performed to clarify the cause of persistent oliguric AKI despite adequate conservative treatment. Light microscopic examination showed extensive interstitial inflammation with a massive infiltration of histiocytic cells without Michaelis-Gutmann bodies. Special staining showed CD68 positivity in infiltrated histiocytes, Von Kossa (for calcium) negativity, and Prussian blue (for iron) negativity. Additionally, C1q staining and electron dense deposits were shown in mesangial matrix without clinical and serological evidence of systemic lupus erythematosus. The final pathologic diagnosis was megalocytic interstitial nephritis accompanied by C1q nephropathy (Fig. 1).
Intravenous administration of methylprednisolone 1 mg/kg was initiated for megalocytic interstitial nephritis and severe infection-related hemophagocytic lymphohistiocytosis that was seen in the histological examination of the patient's kidney and bone marrow, respectively. High-dose steroid treatment was performed for one week, and steroid was halved every 3 days for 2 weeks. Both oliguric AKI and severe leukocytosis were dramatically improved after steroid treatment (Fig. 2).
After using steroids for 12 days, her kidney function had improved enough to stop dialysis. On the day of discharge, her serum creatinine level was 2.48 mg/dL. Her renal function had improved further by her first visit to the outpatient clinic; the serum creatinine level was 1.95 mg/dL.
DISCUSSION
Megalocytic interstitial nephritis is an uncommon form of interstitial nephritis affecting mainly the renal cortex in an otherwise normal kidney. This disease was first described by Zollinger in 1945 (4). The diagnosis of megalocytic interstitial nephritis must be histologically distinguished from two other inflammatory conditions: renal parenchymal malakoplakia and xanthogranulomatous pyelonephritis (5). All of these conditions represent histological variant expression of chronic inflammation and are associated with Gram-negative bacterial infection (4, 6). Although inflammation is caused by infiltration by various inflammatory cells, histiocytes (a type of immune cell that eliminates foreign materials as a part of the host defense) were reported to play a key role in the pathogenesis of megalocytic interstitial nephritis (6). The pathogenic mechanism is suspected to be associated with impairment of bacterial clearance by neutrophils and macrophages, especially in immunodeficient patients (7). Alcohol abuse, as in our patient, was reported as another risk factor of this disease. Göttz et al. (8) previously reported an alcoholic patient with E. coli bacteremia and biopsy-proven megalocytic interstitial nephritis. In that paper, chronic alcohol consumption was reported to cause immune system damage and subsequently facilitate the development of the disease.
There is no clear clinical distinction between megalocytic interstitial nephritis and malakoplakia (9). Megalocytic interstitial nephritis might be an early stage or a morphologic variation of malakoplakia. Malakoplakia is an uncommon form of chronic inflammatory granulomatous disease that most frequently affects the urinary tract. Various organs such as the genitourinary tract, skin, retroperitoneum, lung, gastrointestinal tract, testis, thyroid, and bone also can be involved (10). Kidney parenchymal involvement was reported in only 15% of patients with malakoplakia (3). The typical histologic finding of renal malakoplakia shows many histiocytes with Michaelis-Gutmann (MG) bodies, a distinctive basophilic inclusion containing calcium, phosphate, and often iron. These MG bodies are indicative of malakoplakia and will stain with von Kossa stain (for calcium), Perls' stain (for iron), Prussian blue (for iron), and alizarin red (for calcium) (11). The histology of megalocytic interstitial nephritis also reveals a polymorphous cellular infiltrate with predominant histiocytes containing crystalloid material called von Hansemann cells but no MG bodies (12). The histological features were compatible with megalocytic interstitial nephritis. The coexistence of C1q nephropathy which is characterized by dominant or codominant C1q staining (≥2+ intensity) primarily in the mesangium was also reported. Because C1q molecules have affinity to monocytes, macrophages, and lymphocytes, and C1q receptors are present in the mesangial matrix, the attachment of C1q molecules to histiocytes abundant in megalocytic interstitial nephritis may lead to the co-existence of C1q nephropathy in our case (13, 14).
Our patient required hemodialysis for one month and had poor renal function recovery despite clearing the bacteria. The reason for the delayed recovery of renal function was probably a systemic infection so severe that hemophagocytic histiocytes were seen in the bone marrow. Despite appropriate use of antibiotics, serious systemic inflammation as shown by hemophagocytic histiocytosis and megalocytic interstitial nephritis could not be controlled, and thus steroids were used. However, there are no established steroid treatment regimens in megalocytic interstitial nephritis or renal malakoplakia. Jo et al. (15) used methylprednisolone 500 mg/day from the second hospital day, and Al-Sulaiman et al. (5) prescribed pulse methylprednisolone daily for three days after admission. In these two patients, a high-dose steroid was administered early in the disease course, and the prognosis was excellent. Our patient also was given high-dose steroids late in the disease course and she eventually achieved renal functional improvement. Prompt and sufficient use of appropriate antibiotics is the most important treatment for megalocytic interstitial nephritis. Also, steroid administration is worthwhile with regard to prevention of interstitial inflammation progression (16).
In summary, megalocytic interstitial nephritis is difficult to diagnose without histologic examination. Therefore, megalocytic intersitital nephritis should be considered in patients with poor recovery from acute kidney injury following urinary tract infection.

 XML Download
XML Download