

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Although chromosomal aberrations demonstrate important prognostic impacts in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS), a significant proportion of patients do not demonstrate cytogenetic aberrations on metaphase cytogenetic (MC) analysis (1, 2). In addition, patients with similar cytogenetic aberrations demonstrate heterogeneous clinical characteristics. Although molecular genetic assays have revealed numerous genetic aberrations, such as internal tandem duplications (ITD) in FLT, and CEBPA and NPM1 mutations, clinical heterogeneity among patients with similar MC results (especially those with normal cytogenetics) has not been fully elucidated (3, 4, 5, 6, 7).
Despite the established clinical implications of MC, several limitations are also apparent: low resolution, reliance on cell division, and the inability to detect copy-neutral genetic aberrations. Array comparative genomic hybridization (CGH) does not require cell division and demonstrates much greater resolution than MC, allowing for a more accurate quantification of copy number alterations (CNAs) in malignant cells. Another novel array-based technique-single-nucleotide polymorphism (SNP) microarray-can detect copy-neutral loss of heterozygosity (CN-LOH), which reportedly plays a role in oncogenesis by duplicating oncogenes, inhibiting tumor suppressors, and stimulating improper epigenetic programming, and has been recently described in myeloid malignancies (8, 9).
Array CGH and SNP microarray have been used to study MDS, myeloproliferative disorders (MPD), and AML (mainly in adult patients), and new cryptic lesions have been detected in many patients with normal or abnormal karyotypes using MC analysis (10, 11, 12). Furthermore, previously cryptic CNAs and CN-LOH reportedly demonstrate prognostic significance in adult AML and MDS (10, 11). However, whole-genome scanning using array CGH and SNP microarray are insufficient for studying pediatric AML and MDS, and their clinical and biological implications still need to be determined for use in pediatric populations.
The combination array CGH plus SNP microarray (CGH+SNP microarray) platform can simultaneously detect CNAs and CN-LOH at high-resolution without running 2 separate microarray experiments. In our present study, we report the simultaneous detection of cryptic CNAs and CN-LOH using CGH+SNP microarray and characterize the clinical significance of these submicroscopic defects in childhood AML and MDS.
MATERIALS AND METHODS
Patients
In total, 18 patients with AML (n=15) or MDS (n=3) who were <18 yr at the time of diagnosis were included in this study. All patients were diagnosed between 2008 and 2011 at Asan Medical Center. DNA was extracted from frozen bone marrow (BM) (n=17) or peripheral blood (PB) samples (n=1) that were collected at diagnosis (n=15) or relapse/persistence (n=3) and used in subsequent CGH+SNP microarray analyses.
The clinical characteristics of the study population are listed in Table 1. The median age at diagnosis was 11 yr (range: 0.5-17 yr), and 7 male and 11 female patients were included in our cohort. Patients were categorized according to the World Health Organization (WHO) classification system (13). Nine patients having AML with recurrent genetic abnormalities, 2 patients having AML with myelodysplasia-related changes (AML with MDS), 4 patients having uncategorized AML, and 3 patients having MDS were included.
The clinical characteristics and treatment outcomes of our patients are summarized in Table 2. In the AML group (n=15), 6 patients remained alive and categorized as in complete remission (CR) after receiving allogeneic hematopoietic stem cell transplantation (HSCT), 2 patients were alive and in CR following the administration of only chemotherapy, 3 patients died of relapse after receiving HSCT, 1 patient died of pulmonary hemorrhage after receiving HSCT, and 3 patients died of refractory disease without receiving allogeneic HSCT. In the MDS group (n=3), 2 patients were alive and categorized as in CR after receiving allogeneic HSCT and 1 patient died of graft-versus-host disease and fungal infection after receiving allogeneic HSCT. In total, after a median follow-up period of 28 months (range: 8-63 months), 10 patients are alive and categorized as in CR and 8 patients are dead. The median survival time was 15 months (range: 6-62 months).
Cytogenetic and molecular analyses
All 18 patients in the present study series were successfully assessed using MC analysis. G-banded karyotyping was performed using standard techniques. Whenever possible, 20 metaphases were analyzed. Chromosomal abnormalities are described according to the International System for Human Cytogenetic Nomenclature (14). Fluorescence in situ hybridization (FISH) analysis was performed if requested by the clinician.
RT-PCR was performed on 15 AML patients to detect fusion gene transcripts, including RUNX1-RUNX1T1, PML-RARA, CBFB-MYH11, BCR-ABL, and MLL-AF4. Notably, 7 AML patients were analyzed using multiplex RT-PCR (HemaVision; Bio-Rad Laboratories, Hercules, CA, USA), which can detect 28 different leukemia-associated translocations. c-KIT mutation analysis was performed on 2 patients with the RUNX1-RUNX1T1 fusion transcript. FLT3 was analyzed in 14 AML patients to detect the FLT3-ITD and FLT3-D835 mutations. The NPM1 mutation was analyzed in 5 patients with AML.
Array CGH and SNP microarray analysis
Genomic DNA (gDNA) was isolated from frozen BM (n=17) or PB samples (n=1) using the QiagenDNeasy Blood & Tissue Kit (P/N 69504;Qiagen Inc., Valencia, CA, USA) and processed according to the manufacturer's protocol for the Agilent SurePrint G3 Cancer CGH+SNP 4×180K microarray (P/N G4869A; Agilent Technologies, Santa Clara, CA, USA). All samples were processed according to the instructions for Agilent's oligonucleotide array-based CGH for gDNA analysis and the enzymatic labeling of blood, cells, or tissues (high-throughput option; version 6.3; P/N G4410-90010).
gDNA was digested using the AluI and RsaI restriction endonucleases, which enable the identification of SNPs located within the enzymes' recognition sites. Experimental samples were labeled with cyanine 5-conjugated dUTP and hybridized to the SurePrint G3 Cancer CGH+SNP 4×180K microarrays in the presence of Cot-1-DNA (Invitrogen, Carlsbad CA, USA) for 24 hr at 65℃. A reference HapMap sample with a known genotype (European male; NA 12891_v1) was labeled with cyanine 3-conjugated dUTP and hybridized to the same microarrays. The microarray slides were scanned at 3-µm resolution using an Agilent microarray scanner, and images were extracted using Agilent Feature Extraction software (v10.10) that was integrated in Agilent CytoGenomics 2.0.6.0. Data were analyzed using Agilent CytoGenomics 2.0.6.0 with an Aberration Detection Method-2 (ADM-2) algorithm (threshold 8.0). Results were analyzed according to genome build Hg19.
Statistical analysis
The Kaplan-Meier method was used to calculate survival outcomes. Outcomes were compared using the log-rank test. In this study, P<0.05 is considered statistically significant. All statistical analyses were performed using SPSS (Statistical Package for the Social Science, version 18.0; IBM, Somers, NY, USA).
RESULTS
Conventional cytogenetics
In the AML group (n=15), MC analysis revealed normal karyotypes in 3 patients and chromosomal aberrations in the other 12 patients, including balanced translocation (11 translocations in 10 patients) and/or chromosomal CNAs such as numerical chromosomal abnormalities and/or the duplication or deletion of chromosomal segments (3 gains and 6 losses in 7 patients). In the MDS group (n=3), MC analysis revealed normal karyotypes in 2 patients and balanced translocation and two chromosomal additions was noted in the other 1 patient. In total, MC analysis revealed CNAs in 8 patients. These results are summarized in Table 2 and Table 3.
Molecular genetic abnormalities
RT-PCR analysis demonstrated RUNX1-RUNX1T1 with c-KIT mutation (n=1), wild-type c-KIT (n=1), CBFB-MYH11 (n=1), PML-RARA (n=1), MLL-AF10 (n=1), MLL-AF6 (n=1), and DEK-NUP214 (n=2), which is consistent with conventional cytogenetic analysis. Fourteen patients with AML were analyzed for the FLT3 mutation, and 3 patients demonstrated the FLT3-ITD mutation. None of these 3 patients demonstrated the NPM1 mutation. One of these 3 patients with the FLT3 mutation demonstrated a normal karyotype, whereas 2 patients demonstrated t(9;11) and t(6;9), respectively. These results are shown in Table 2.
CNA identification
The CGH panel (Agilent SurePrint G3 Cancer CGH+SNP 4×180K microarrays) detected 14 CNAs (7 gains and 7 losses) in 9 of 18 patients (50%), while conventional cytogenetics detected 11 CNAs (5 gains and 6 losses) in 8 of 18 patients (44%; Table 3). All genomic imbalances detected using MC analysis were recognized by CGH, whereas 2 gains and 1 loss detected by CGH were undetected by MC analysis. In addition, an accurate gain region that was erroneously detected by MC was corrected using CGH (patient 8). Moreover, CGH more accurately detected breakpoints. In patient 6, CGH detected an accurate breakpoint in derivative chromosome 12, including the loss of 12p13.33-p12.3 (Fig. 1A). In 11 patients with balanced translocations, CGH did not detect any alterations in the breakpoints of the translocated chromosome. Recurrent segmental gain or loss was not identified.
The cryptic CNAs detected using CGH include the following. In patient 2 with 45,X,-Y,t(8;21)(q22;q22), CGH detected additional small deletions in 1q42.13-q42.2 (2.24 Mb). In patient 8, who demonstrated 47,XX,t(6;9)(p23;q34),+add(21)(q21) according to MC analysis, CGH revealed that the addition of 21q21 was actually the gain of 13q21.32-q34. In patient 14, initial BM examination revealed acute myelofibrosis without evidence of hematological malignancy. Using the samples collected during the initial BM examination, MC analysis demonstrated a normal karyotype whereas CGH detected trisomy 8 and 19. Subsequent BM examination performed 2 months later using MC revealed acute megakaryoblastic leukemia with 54,XX,+2,+6,+7,+8,+8,+19,+19,+22. Table 2 summarizes all of the CNAs detected using MC and CGH.
LOH identification
Using Agilent SurePrint G3 Cancer CGH+SNP 4×180K microarrays, LOHs>5 Mb were detected in 12 regions in 8 of 18 patients (44%), and LOHs>10 Mb were detected in 6 regions in 5 of 18 patients (28%). Of 12 LOHs, 9 LOHs were involved in interstitial segments, 2 in terminal regions, and 1 in whole chromosomes. Thus, LOHs>10 Mb that involved terminal regions or whole chromosome were detected in 3 regions in 3 of 18 patients (17%; Table 2). Of 12 LOHs, 1 LOH was a copy-gain LOH involving trisomy 6 and the others were CN-LOHs. Three of 8 patients demonstrated>1 LOH. LOH affected the chromosomal arms of 1p, 5q, 9p, 7q, 13q, 6,15q, 18q, Xp, and Xq. Recurrent LOH was also observed at 1p33-p32 (n=2).
Notably, of 3 patients with the FLT3-ITD mutation (patients 5, 9, and 10), patient 9 demonstrated LOH 13q-harboring FLT3, which resulted in a high allelic ratio of 1.847; however, patient 5 did not demonstrate LOH and patient 10 demonstrated a small LOH at 7q. Patient 9 achieved delayed CR after 3 cycles of induction chemotherapy and experienced early relapse. This patient subsequently died of refractory disease.
Among patients with acute basophilic leukemia (patients 6 and 15), only patient 6 demonstrated LOH at Xq22, and survived in CR after receiving allogeneic HSCT. Patient 15 demonstrated 2 LOHs involving 1p33-p32 (described above as recurrent LOH) and 9pter-p13, which is a telomeric defect>30 Mb (Fig. 1B). Notably, patient 15 demonstrated the aberrant expression of lymphoid antigens (CD10 and CD19) at diagnosis. Patient 15 demonstrated early relapse during consolidation chemotherapy and subsequently died of refractory disease.
MC analysis determined trisomy 6 in patient 12, which resulted from LOH and concomitant isochromosome formation according to SNP microarray analysis (Fig. 1C). Patient 12 relapsed 8 months after receiving allogeneic HSCT and received reinduction chemotherapy followed by a second round of allogeneic HSCT.
Comparison of CGH+SNP microarray and MC analyses
Table 3 compares MC and CGH+SNP microarray analysis for the detection of cytogenetic defects. MC detected 11 CNAs in 8 patients, while CGH+SNP microarray revealed 14 CNAs in 9 patients as well as 12 LOHs in 8 patients. Of 5 patients with normal karyotypes (patients 10, 14, 15, 16, and 17), CGH+SNP microarray revealed LOH in 2 patients (patients 10 and 15) and LOH and genomic gain in 1 patient (patient 14), revealing that 3 patients actually had genetic abnormalities. Of 13 patients with abnormal MC, CGH+SNP microarray detected additional, previously cryptic CNAs (n=2) and LOHs (n=5) in 6 patients. In total, 9 patients demonstrated additional aberrations, including CNAs (n=3) and/or LOHs (n=8).
Clinical impact of lesions identified using CGH+SNP microarray
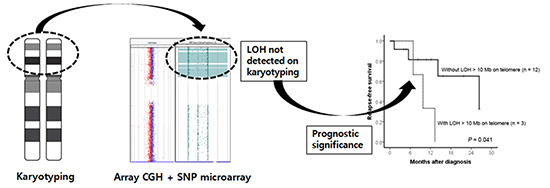
Of 5 patients with normal karyotypes determined using MC analysis, 2 patients without hidden genetic abnormalities survived after receiving allogeneic HSCT. Contrarily, 3 patients with LOH and/or CNA detected using CGH+SNP microarray died of refractory disease (patients 10 and15) or pulmonary hemorrhage following allogeneic HSCT (patient 14). All 3 patients with LOH>10 Mb involving the terminal region or whole chromosome demonstrated relapse (patient 12) or disease progression (patients 9 and15; Table 2). Analyzing the impacts of LOH on the survival outcome in 15 patients with AML, 3 patients with terminal LOH>10 Mb showed significantly inferior relapse-free survival (RFS) rate (P=0.041). Overall survival did not differ between patients with or without terminal LOH>10 Mb (Fig. 2).
DISCUSSION
In this study, genetic aberrations were analyzed in children with AML or MDS using high-resolution genome-wide CGH+SNP microarray. Our results show that CGH+SNP microarray not only confirms the CNAs detected by MC analysis but also simultaneously identifies cryptic CNAs and CN-LOHs. CGH+SNP microarray detected more CNAs than MC analysis (14 CNAs in 9 patients vs. 11 CNAs in 8 patients) and revealed LOHs that were not detected by conventional cytogenetic analysis in 8 of 18 patients (44%). Notably, 3 of 5 patients with normal karyotypes demonstrated cryptic genetic aberrations, which underscores the genetically heterogeneous nature of cytogenetically normal AML. Moreover, CGH+SNP microarray facilitated the detection of additional previously cryptic abnormalities in patients with abnormal karyotypes and determined the exact sites of chromosomal abnormalities, which were erroneously detected by MC analysis.
Previous studies show that SNP microarray and oligonucleotide CGH can more sensitively detect chromosomal abnormalities than MC analysis in AML and MDS patients (15, 16, 17). Cytogenetically normal AML (17, 18), as well as specific AML subtypes such as AML with complex karyotypes (19), core-binding factor AML (20), acute promyelocytic leukemia (21), and secondary AML (15), reportedly demonstrate hidden genetic alterations that can be detected using array CGH and/or SNP microarray. In our present study, patients within the same WHO classification, as well as those with normal karyotypes, demonstrated individually different, additional cryptic abnormalities according to CGH+SNP microarray. These results imply that clinical heterogeneity attributes to genetic aberrations in patients with similar MC results, especially patients with normal karyotypes.
The clinical implications of CGH and SNP microarray analyses have been evaluated in previous studies, which report the potential prognostic impact of previously cryptic CNAs and CN-LOHs (15, 16). However, because prognostic values were evaluated in various patient populations by diverse studies, the clinical applications of such results are still limited. In normal-karyotype MDS, patients with cryptic genomic imbalances demonstrate inferior survival (16). Similarly, patients with SNP microarray lesions, including acquired somatic CN-LOH, exhibit a poorer overall survival and event-free survival than patients with primary AML and normal MC (15). It is notable that among 5 of our patients with normal MC, 3 patients with cryptic genetic aberrations did not survive while 2 patients without cryptic aberrations remain alive and relapse-free. Moreover, all 3 of our patients with large, terminal LOHs demonstrated significantly inferior RFS. These observations may imply the prognostic significance of cryptic genetic aberrations. However, it was difficult to evaluate the significance of the prognostic impact of hidden CNAs and CN-LOHs in our study population as it was small and heterogeneous. Thus, prognostic implication of cryptic genetic aberrations should be investigated in a larger homogeneous population.
In our present study series, LOHs>5 Mb were detected at 12 regions in 8 of 18 patients (45%), and LOHs>10 Mb were detected in 6 regions in 5 of 18 cases (28%). This study, however, did not include matched DNA controls because of its retrospective design, and LOH frequency was most likely overestimated. When more stringent LOH size and location criteria were applied, LOHs>10 Mb involving the terminal regions or whole chromosome were detected in 17% of patients. This is consistent with previous studies reporting that de novo AML is characterized by a low burden of genomic alterations in comparison with pediatric acute lymphoblastic leukemia (ALL) (22).
The detection of recurrent somatic LOH has contributed to the identification of novel mutations associated with myeloid oncogenesis, such as JAK2 in MPD (23) and TET2 (24) and FZH2 (25) in AML. In addition, recurrent somatic LOH indicates a mechanism by which leukemic cells can increase the dose of the mutated gene (15). The individual cases in this study may suggest a mechanism whereby genetic mutations in known oncogenes or suppressors can increase, contributing to leukemogenesis and phenotypic variations in same subtypes.
Patient 9 demonstrated LOH 13q containing FLT3, which resulted in a high allelic FLT3-ITD ratio. Previous studies report that LOH 13q is associated with homozygous FLT3-ITD mutation; thus, the presence of LOH 13q may be linked to clinical outcomes in patients with FLT3-ITD mutation (26).
Patient 12 also demonstrates an important revelation regarding leukemogenesis. Trisomy 6 has been reported as a nonrandom primary numerical anomaly in myeloid disorders that abrogates the usual favorable prognosis associated with RUNX1/RUNX1T1 in AML patients (27, 28). As putative candidate genes, IRF4 and DEK in 6p25.3 and 6p22.3, respectively, may be associated with myeloid leukemogenesis and prognosis in AML with trisomy 6. The role of trisomy 6 in leukemogenesis and its impact on prognosis can be hypothetically attributed to increased oncogenes in chromosome 6, which is caused by LOH and concomitant isochromosome formation. LOH in chromosome 6 in AML patients with trisomy 6 has not been described in other studies. This implies that leukemogenesis in other trisomy karyotypes could be explained by the imprinting effects of certain genes due to acquired trisomy-caused LOH.
This study included 2 patients with acute basophilic leukemia (patients 6 and 15). CGH+SNP microarray revealed different genetic profiles that may underlie differences in phenotypes and genetic aberrations. Patient 6 demonstrated MLL rearrangement and LOH Xq22.1, which harbors BEX1. Interestingly, BEX1 expression levels in MLL-rearranged AML cells are higher than in wild-type-MLL AML cells, and BEX1 could play a role in AML leukemogenesis and MLL rearrangements (29). In addition, CGH demonstrated the loss of ETV6-harboring 12p13.3 in patient 6. The loss of ETV6 is reportedly associated with acute basophilic leukemia (30). Patient 15 demonstrated LOH 9p, which harbors JAK2, CDKN2A, and PAX5. CDKN2A reportedly influences the risk of developing ALL (31). PAX5 is also reportedly concurrently deleted in CDKN2A deletion in B-lineage ALL (32). Segmental LOH 9p could explain this concurrent deletion. It is notable that patient 9 demonstrated the aberrant expression of lymphoid antigens. This biphenotypic feature may be associated with LOH 9p and the harboring of tumor suppressors, which reportedly impact the risk of developing ALL.
In summary, our present findings show that CGH+SNP microarray is superior to conventional cytogenetic analysis for detecting genetic aberrations (other than balanced translocations) in childhood AML and MDS. CGH+SNP microarray simultaneously detects previously cryptic CNAs and LOHs, thereby decreasing the reported proportion of patients with a normal karyotype. Moreover, segmental LOHs harboring oncogenes or tumor suppressors associated with leukemogenesis may implicate pathogenic and prognostic significance. In order to refine the prognostic significance and delineate the pathogenic role of hidden genetic aberrations in childhood AML and MDS, a prospective study on a larger pediatric population with matched DNA samples is needed.





 XML Download
XML Download