

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Metabolic acidosis is associated with the progression of chronic renal failure (CRF) and can be a common consequence of CRF (1). Conversely, many clinical studies have shown that metabolic acidosis itself may be a cause of renal disease progression and that alkali therapy ameliorates its progression (2, 3). In experimental studies, several mechanisms for the effects of alkali therapy have been evaluated, such as the reduced activation of the alternative complement pathway (4), decreased cell proliferation and transdifferentiation (5), and the endothelin (ET) antagonism pathway (6). However, there are few reports of the role of renal acid-base transporters during alkali therapy in remnant kidney models.
Luminal Na/H exchanger type 3 (NHE3) is responsible for a significant portion of renal Na+ and HCO3- absorption (7). NHE3 is variably expressed depending on the pathologic condition and was increased in adriamycin-nephropathy models (8). NHE3 activation plays a role in the pathogenesis of ischemic acute renal failure (9). Under alkali loading, apical NHE3 expression in the kidney can be down-regulated (10). The inhibition of NHE3 may improve kidney function and structure in acute ischemic kidney injuries (11). However, there are few reports of the effects of alkali therapy on apical NHE3 expression in chronic kidney failure. We evaluated the beneficial effects of dietary sodium bicarbonate in ameliorating renal disease progression and the role of NHE3 with regard to these effects in rats with a remnant kidney.
MATERIALS AND METHODS
Experimental animals
Twenty-three specific pathogen-free male Sprague-Dawley rats (5-6 weeks old, 160-190 g; Orient Bio Inc., Seongnam, Korea) were housed with free access to water. Experimental CRF was induced by the excision of approximately two-thirds of the left kidney and a right total nephrectomy. After a 5/6 nephrectomy, all rats were randomly allocated to the NaCl-treated group or the NaHCO3-treated group. CRF rats in the NaHCO3-treated group were given a 20% casein diet with 174 µM/g NaHCO3 (DYET #113316, Dyets Inc., Bethlehem, PA, USA) and were considered alkali-treated animals. CRF rats in the NaCl-treated group were given a 20% casein diet with 174 µM/g NaCl (DYET #113811, Dyets Inc.) and were considered control animals treated with the same amount of dietary sodium. The NaHCO3 and NaCl amounts were determined according to the previous report (6).
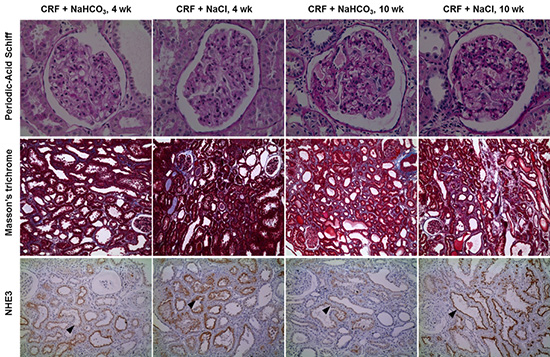
CRF rats in the two groups were euthanized at the time points of 4 and 10 weeks after the operations (n=10 at 4 weeks and n=13 at 10 weeks). Fragments of remnant kidneys were taken from center areas and divided into two parts. One part was used for protein extraction, and the other for pathology examination. The glomerular sclerosis (GS) index was assessed in 50 glomeruli per rat on periodic-acid Schiff-stained paraffin sections at a magnification of 400 × (12). The tubulointerstitial (TI) lesion indices were determined using a semiquantitative scoring system on Masson's trichrome-stained sections at a magnification of 100 × (5). The pathologic scores for GS and TI were performed in blinded fashion.
Physiologic data
Blood pressure was measured using the tail-cuff plethysmography method (IITC Life Sciences, Woodland Hills, CA, USA). For the final 2 days prior to euthanasia, the animals were placed in metabolic cages, and 24-hr urine samples were collected on the day before euthanasia for the measurement of urine urea nitrogen (UUN), creatinine (Cr), protein, osmolality, Na, K, and ET-1 levels. A blood sample was collected from the abdominal aorta at the time of euthanasia for the measurement of blood urea nitrogen (BUN), Cr, osmolality, and electrolyte levels. Biochemical tests were performed using an automatic chemistry analyzer (Hitachi 7070; Hitachi, Tokyo, Japan). Indirect ion-selective electrode methods were used to obtain electrolyte levels, and the enzyme method was used for measuring total CO2 levels. Plasma and urine osmolality were measured with a cryoscopic osmometer (Osmomat 030-D-M; Gonotec, Berlin, Germany). The glomerular filtration rate (GFR) was calculated using the average of urea clearance and creatinine clearance (7).
Semiquantitative immunoblotting
The remnant kidneys were quickly removed and placed in a chilled isolation solution containing 250 mM sucrose, 10 mM triethanolamine (Sigma, St. Louis, MO, USA), 1 ng/mL leupeptin (Sigma), and 0.1 mg/mL phenylmethylsulfonyl fluoride (Sigma), titrated to pH 7.6. The pieces were homogenized at 15,000 rpm with three strokes for 15 sec using a tissue homogenizer (PowerGun 125; Fisher Scientific, Pittsburgh, PA, USA). After homogenization, the total protein concentration of the homogenate was measured by the bicinchoninic acid protein assay method (BCA Reagent Kit; Sigma) and diluted to 2.05 µg/µL using the isolation buffer solution. The samples were then stabilized by heating to 60℃ for 15 min after adding 1 vol 5 × Laemmli sample buffer/4 vol sample. Initially, loading gels were used with each sample set. Five micrograms of protein from each sample were loaded into individual lanes, run on 12% polyacrylamide-SDS minigels using a Mini Protean III electrophoresis apparatus (Bio-Rad, Hercules, CA, USA) and stained with Coomassie blue dye (G-250, Bio-Rad; 0.025% solution prepared in 4.5% methanol and 1% acetic acid), which were considered internal standards (13). Selected bands from these gels were scanned (GS-700 Imaging Densitometry; Bio-Rad) to determine the density (Molecular Analyst version 1.5; Bio-Rad), and relative amounts of protein were loaded into each lane. Finally, protein concentrations were corrected to reflect these measurements and were subjected to immunoblot analysis, using a previously described method (7).
Immunohistochemistry
The other half of the left kidney from each rat was immersed in a 4% paraformaldehyde solution at 4℃ overnight. Each slice was dehydrated with a graded series of ethanol and embedded in paraffin. The embedded pieces of kidney were sectioned at a 3 µm thickness on a microtome (RM 2145; Leica Instruments GmbH, Nussloch, Germany) and mounted on gelatin-coated glass slides. The sections were deparaffinized with xylene, dehydrated with a graded series of ethanol, and rehydrated. Antigen retrieval was performed by repeated boiling and cooling in a citric acid buffer. Endogenous peroxidase activity was blocked by incubation of the sections in 3% H2O2 for 10 min. Next, the immunostaining procedure was performed according to the protocol of the Dako Cytomation kit (Envision + Dual Link System-HRP; Carpinteria, CA, USA). The sections were incubated for 1 hr with antibody against NHE3 and Na-K-ATPase at room temperature.
Primary antibodies and cytokines assay
For semiquantitative immunoblotting and immunohistochemistry, we used previously characterized polyclonal antibodies. Affinity-purified polyclonal antibodies against NHE3, NKCC2, NCC, ENaC-α, ENaC-γ, NBC, pendrin, and H-ATPase were used as described in a previous study (7, 14). Affinity-purified polyclonal antibodies against Na-glucose cotransporter-1 (SGLT1; AB1352; Millipore Corp., Billerica, MA, USA), ENaC-β (sc-21013; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and Na-K-ATPase (05-396; Millipore Corp.) were also used. Tissue ET-1 concentrations were measured using QuantiGlo ET-1 ELISA (QET00B; R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Statistical analysis
Comparisons between the two groups were made with the Mann-Whitney U-test, and comparisons among three groups were made with the Kruskal-Wallis test (SPSS software; SPSS Inc., Chicago, IL, USA). Band density values were standardized by dividing them by the average value of the control group. The mean for the control group was defined as 100%, and the results were expressed as the means±SEM. P<0.05 was considered statistically significant.
RESULTS
Change of Physiologic and Pathologic Data
After a 5/6 nephrectomy, serum total CO2 levels in the NaHCO3-treated group were higher compared with the control group throughout the study period (Table 1). At 4 weeks, the GFR in the NaHCO3 group was slightly higher than that in the NaCl group, although it was not statistically significant. At week 10, the GFR in the NaHCO3-treated group was significantly higher than that in the control group. GFRs between week 4 and week 10 remained constant in the NaHCO3-treated group, while GFRs decreased in the control group. The degree of proteinuria and the systolic blood pressure were similar at week 4 and week 10 between the groups consuming NaCl and NaHCO3. Fig. 1 shows the dietary Na intake per day (input), urinary Na excretion per day (output), and Na balance (the difference between input and output). There were no differences in the Na balance between the two groups at week 4 and week 10. At week 4, the GS index in the NaHCO3-treated group was less severe compared to the NaCl-treated group (0.17±0.041 vs 0.47±0.063, respectively, P<0.001) (Fig. 2). These differences persisted at week 10 (0.66±0.063 in the NaHCO3-treated group vs 0.99±0.074 in the NaCl-treated group, P=0.001). At week 4, the TI damage index in the NaHCO3-treated group was half of that in the NaCl-treated group (0.45±0.010 vs 0.92±0.120, respectively, P=0.004). Similarly, the TI damage index in the NaHCO3-treated group was 3/4-fold of that in the NaCl-treated group (1.33±0.12 vs 1.80±0.12, respectively, P=0.010). Both GS and TI damage indices gradually progressed between week 4 and week 10 in the two groups.
Change of renal transporters
Fig. 3 shows the immunoblots for renal Na and acid-base transporters. The expression of NHE3 in the NaHCO3-treated group was significantly decreased compared to the control group at week 4 (10.1±4.25 vs 100±21.1, respectively, P=0.007). At week 10, the expression of NHE3 in the NaHCO3-treated group was decreased, compared to the control group; however, the decrease was not statistically significant (37.1±13.0 vs 100±52.9, P=0.308). Immunohistochemistry revealed that apical expression for NHE3 was more prominent in the NaCl-treated group than in the NaHCO3-treated group at week 4 and week 10.
Na-K-ATPase expression in the NaHCO3-treated group was significantly decreased at week 4 compared to the NaCl-treated group (57.8±13.0 vs 100±11.5, respectively, P=0.025). There was no difference in the expression of Na-K-ATPase between the two groups at week 10. On immunohistochemistry, the basolateral expression for Na-K-ATPase was increased in the NaCl-treated group at both week 4 and week 10 (Fig. 4).
There was no change in the densities of NKCC2, SGLT1, H-ATPase, NBC, and pendrin in the two groups at week 4 and week 10. The densities of NCC increased and those of ENaC-α decreased in the NaHCO3-treated animals at week 10.
At week 4, there was no difference in the renal ET-1 levels between the NaHCO3-treated group and the NaCl-treated group (0.18±0.096 vs 0.15±0.019 [pg/mL]/[mg/mL], respectively) (Fig. 5). In contrast, the renal ET-1 levels in the NaHCO3-treated group were decreased at week 10, compared with the NaCl-treated group (0.32±0.15 vs 1.14±0.20 [pg/mL]/[mg/mL], respectively, P=0.021). In addition, the NaCl-treated rats were increased at week 10, compared with the same group at week 4 (P=0.011).
DISCUSSION
Our data show that dietary sodium bicarbonate in the nephrectomized models may have beneficial effects in ameliorating the decrease in GFR and pathologic damage. Our data also show that these effects may be associated with NHE3 expression as well as ET-1 levels. NHE activity in the myocardium is associated with cardiac remodeling (15), and NHE inhibition may lead to the regression of myocardial fibrosis (16). Renal NHE expression was upregulated in adriamycin-induced nephropathy in parallel with the degree of glomerulosclerosis and interstitial fibrosis, and the preventive effects of amiloride on renal lesions suggest the potential importance of NHE (17). The inhibition of NHE may be beneficial for protection in cases of decreased kidney function as well as tubular injury in acute kidney injury (11). This study provides evidence that NHE3 inhibition may be associated with renal protective effects in CRF.
Chronic metabolic acidosis induced by acid loading enhances NHE3 protein abundance and transport activity in the rat thick ascending limb (18). After the correction of metabolic acidosis with sodium bicarbonate in our experiment, NHE3 expression was decreased compared with the control group. NaHCO3 loading can directly downregulate apical NHE3 expression in the rat kidney proximal tubule (10). The downregulation of NHE3 could be responsible for a decreased acid burden due to the correction of metabolic acidosis and increased excretion of alkaline excess in nephrectomized rats subjected to NaHCO3 loading.
In the previous study, we evaluated the expression of renal tubular transporters in 5/6 nephrectomized rats with a regular diet (7). Increased urinary sodium excretion was associated with decreased expression of renal sodium transporters, especially NHE3 in the proximal tubule. There was no difference between the two groups in terms of sodium loading and sodium balance at week 4 and week 10, but NHE expression in the NaHCO3-treated group was decreased more than in the NaCl-treated group. This suggests that the downregulation of NHE3 may be affected by alkali loading independent of sodium loading in CRF. In contrast, the expression of H-ATPase, NBC, or pendrin, which are major regulators of acid-base homeostasis, may not be associated with alkali therapy in CRF rats. Therefore, NHE3 may be a main target of bicarbonate therapy.
Augmented intrinsic acid production promotes TI injury through endothelin receptors (19). Chronic metabolic acidosis induces increased ET expression in the renal proximal tubule (20, 21). Moreover, ET expressed by the kidney can activate proximal tubule acidification by activating the proximal tubule NHE, while ET has a lack of effects on the activities of the apical SGLT (22). This effect of ET has been shown to involve the trafficking of NHE3 to the apical membrane, which is achieved by an increase in the exocytic insertion of NHE3 into the apical membrane (21, 23). In our study, apical membrane NHE abundance was decreased in the alkali-treated group with relatively decreased ET levels.
Chronic metabolic acidosis may stimulate NKCC2 of the rat medullary thick ascending limb (24). In our data, NKCC2 expression was not different between the NaHCO3- and NaCl-treated groups. This may be related to the decreased expression of NHE3, which may be associated with increased distal sodium delivery. NH4Cl-induced metabolic acidosis in rats is associated with a strong downregulation of NCC in the distal convoluted tubules (25). In our data, NCC expression tended to be increased in the NaHCO3-treated group, compared with the NaCl-treated group. It is possible that the expression of NCC, decreased in acidic condition, may be restored under the alkali therapy.
When NHE3 was stimulated by feeding NH4Cl, Na-K-ATPase activity was increased (26). When NHE3 was inhibited by the administration of amiloride, however, Na-K-ATPase activity was decreased in rat kidney proximal convoluted tubule segments (26). In our data, Na-K-ATPase expression was decreased at week 4, and the basolateral reactivity of the Na-K-ATPase was also decreased at week 4 and week 10 in the NaHCO3-treated group. The change of Na-K-ATPase activity may be affected by altered NHE3 expression.
Acidification inhibits ENaC activity, whereas alkalinization stimulates ENaC (27). In our data, most ENaC abundance was not changed in both groups, although α-ENaC density was decreased in the NaHCO3-treated rats at week 10. In rats with normal renal function, NaHCO3 or NaCl loading downregulates basolateral NBC (10). In our nephrectomized rats, NaHCO3 loading did not affect NBC expression. In addition, the expression of pendrin or H-ATPase in the distal tubule did not change through the correction of acidosis. It is possible that renal acid-base transporters under CRF conditions could be regulated by a different mechanism. The similar expression of pendrin in two CRF groups may lead to ENaC, because pendrin modulates ENaC abundance under luminal alkalinization (28).
Phisitkul et al. (6) demonstrated the changes of GFR in 5/6 nephrectomized rats with a variety of diet and alkali therapy. The NaHCO3 treatment that was under the control of blood pressure showed better GFR outcomes than NaHCO3 treatment alone. In our study, we did not give any anti-hypertensive medication to the two groups. In both groups, high blood pressures may promote GS and/or TI damage and altered expression of renal tubular transporters, although there was no difference in the blood pressure in the two groups. Further experiments on the role of renal transporters in CRF rats under the control of blood pressure should be performed.
In summary, dietary sodium bicarbonate intake contributed to decrease of NHE3 and ET levels. Expression of Na-K-ATPase and NCC in CRF rats may be directly or indirectly related to the alteration of NHE abundance. This finding suggests that alkali treatment in CRF may have beneficial effects in ameliorating renal disease progression, and these effects may be mainly associated with the altered expression of NHE3 in the kidney, which may be a therapeutic target for renal disease progression.






 XML Download
XML Download