

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are fatal neurodegenerative disorders that affect humans and animals. These diseases are characterized by spongiform changes, astrogliosis, and the accumulation of an abnormal prion protein (PrPSc) in the central nervous system (CNS). The key mechanism in the pathogenesis of prion diseases is the conversion of the cellular prion protein (PrPC) into PrPSc (1). The human prion diseases include kuru, Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI) (2). The majority of human prion diseases are sporadic (85%). Approximately 10%-15% of human prion diseases are inherited, i.e., caused by mutations in the prion protein gene (PRNP), and less than 1% are acquired (3, 4).
PRNP is located on chromosome 20p12 in humans. The human PRNP gene contains two exons, and the 253 amino acid prion protein (PrP) is encoded by the larger second exon (5). PrP is an N-linked glycosylated protein that is posttranslationally processed to remove a 22 amino acid signal peptide and is attached to the cell membrane by a glycosylphosphatidylinositol (GPI) anchor. The N-terminal domain of human PrP comprises a five octapeptide repeat, and the C-terminal domain contains two N-glycosylation sites and an intermolecular disulfide bond (6).
To date, more than 30 mutations of PRNP have been found in the open reading frame (ORF) of this gene (3, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30). These mutations are the only cause of familial prion diseases, which include familial CJD, GSS, and FFI (31, 32, 33). In addition to these mutations, many polymorphisms have also been observed in the ORF of PRNP (3, 34). In particular, single nucleotide polymorphisms (SNPs) at codons 129 or 219 of PRNP represent susceptibility factors for human prion diseases (35, 36, 37). Candidate gene studies and genome-wide association studies (GWAS) have been conducted to identify genetic susceptibility factors for human prion diseases (38, 39, 40).
In this review, we summarize the genetics of familial human prion diseases and current studies of the genetic factors in sporadic human prion diseases.
PRNP MUTATIONS
Genetic CJD
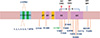
Familial CJD is caused by inherited autosomal dominant point mutations and insertion/deletion mutations of octapeptide repeats (OPRI/OPRD) (41). Among these mutations, many have been identified in patients without a family history of prion disease, known as genetic CJD. Genetic CJD accounts for 5%-15% of all CJD cases. Genetic CJD may be caused by point mutations at codons 114 (GGT→GTT), 178 (GAC→AAC), 180 (GTC→ATC), 183 (ACA→ACG), 188 (ACG→AAG), 196 (GAG→AAG), 200 (GAG→AAG), 203 (GTT→ATT), 208 (CGC→CAC), 210 (GTT→ATT), 211 (GAG→CAG), 232 (ATG→AGG), or 238 (CCA→TCA), or by insertional mutations of 1, 2, 3, 4, 5, 6, or 7 octapeptide repeat segments (Table1 and Fig. 1) (2, 3, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18).
The distribution and frequency of PRNP mutations in genetic CJD differ between Europeans and East Asians (Table 2). The most common PRNP mutation in European genetic CJD patients is in the codon 200, followed by mutations in codons 210 and 178, whereas the most common mutation in Japanese CJD patients is in codon 180, followed by mutations in codon 200 and 232 (42, 43). In particular, the PRNP mutation at codon 210 is prevalent in European countries but rare in East Asian populations. Conversely, that mutation in codon 232 has been observed in Japanese patients but not European patients. The frequency of mutations in codons 180 (P<0.001), 200 (P<0.001), 210 (P<0.001), and 232 (P<0.001) are significantly different between Europeans and East Asians. However, the frequencies of point mutations in codons 171, 178, 188, 196, 203, 208, and 211 were not significantly different between Europeans and Asians. Several mutations in the ORF of PRNP have been found in genetic CJD patients in Korea. Five mutations in codon 180, three in codon 200, two in codon 203, and two in codon 232 have been identified (44, 45, 46, 47, 48, 49). The frequency of genetic CJD in Korea is very similar to that in Japan.
The onset of genetic CJD typically occurs between 30 and 55 yr of age with progressive confusion and memory impairment, ataxia, and myoclonus. The duration of genetic CJD ranges from a few months to several years (50). The mean age at disease onset and the mean duration in genetic CJD was linked to methionine (Met) or valine (Val) at codon 129 in the mutated allele. PrPSc type, histological changes, and clinical features of genetic CJD patients with most mutations in cis with Met at codon 129 were overall very similar to those of the sporadic CJD MM1 phenotype.
GSS
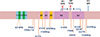
GSS has been associated with point mutations at codons 102 (CCG→CTG), 105 (CCA→CTA, ACA, TCA), 117 (GCA→GTG), 131 (GGA→GTA), 145 (TAT→TAG), 160 (CAA→TAA), 187 (CAC→CGC), 198 (TTC→TCC), 202 (GAC→AAC), 211 (GAG→GAC), 212 (CAG→CCG), 217 (CAG→CGG), 226 (TAC→TAA), and 227 (CAG→TAG), and insertional mutations of 8 and 9 octapeptide repeat segments (Table 1 and Fig. 2) (2, 3, 7, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29).
The distribution and frequency of PRNP mutations in GSS were also clearly distinct between Europeans and East Asians (Table 2). The most common PRNP mutation in GSS patients in European countries and East Asia is in codon 102. The PRNP mutation at codon 105 is observed in East Asian, but not European populations. In contrast, the mutation in codon 117 is found in European, but not East Asian populations (42, 43). There were significant differences in the frequencies of three mutations (codon 102, P<0.001; codon 105, P=0.018; codon 117, P=0.019) between European and East Asian GSS patients. In Korea, a mutation at codon 102 of PRNP has been reported in two GSS patients (44, 51).
The onset of GSS mainly occurs between 40 to 60 yr of age. The clinical symptoms of GSS include cerebellar dysfunction, gait disturbance, dementia, and mild dysarthria. All GSS cases exhibit PrP plaque deposits (50). The hallmark of GSS is the extensive PrP-amyloid deposits with minimal spongiform change. In addition, neurofibrillary tangles have been detected in the GSS patients with PRNP mutations at codon 105, 145, and 217 (52, 53, 54).
FFI
FFI is caused by a mutation at codon 178 (GAC→AAC) of PRNP in combination with a polymorphism that generates a Met at codon 129 (Table 1 and Fig. 1) (2, 3, 7, 34). The frequency of the PRNP mutation at codon 178 in conjunction with M129 is more prevalent in European than East Asian countries (P<0.001) (Table 2) (42, 43). In Korea, a mutation at codon 178 accompanied by M129 has been reported in one FFI patient (46).
FFI typically presents between 20 and 72 yr of age, with an average age of onset of approximately 50 yr. The duration of FFI ranges from 6 months to 33 months with an average of 18.4 months. The major clinical symptom of FFI is insomnia (50). Ataxia, dysarthria, myoclonus, dysphagia and pyramidal signs can also be observed.
PRNP POLYMORPHISMS
In addition to the mutations described above, many polymorphisms have been observed in the ORF of PRNP. PRNP polymorphisms are observed at codons 129 (ATG→GTG), 142 (GGC→AGC), 171 (AAC→AGC), 188 (ACG→AAG), and 219 (GAG→AAG), and the deletion of 1 octapeptide repeat segments is also considered a polymorphism (2, 3, 55).
Codon 129 SNP
The PRNP codon 129 SNP introduces an amino acid substitution of Val for Met. The SNP at codon 129 of PRNP has been considered a genetic risk factor for human prion diseases (34, 37). This SNP was strongly associated with sporadic CJD in Korean, Japanese, Dutch, British, Spanish, French and German populations (Table 3) (37, 38, 43, 55, 56, 57, 58, 59, 60, 61, 62). Heterozygosity at codon 129 is protective against sporadic, iatrogenic or variant CJD in Europeans and East Asians (35, 37, 38, 58, 59, 60, 61, 62, 63, 64, 65). In particular, all cases of variant CJD are homozygous for Met at this SNP (65). The frequency of Met homozygosity at codon 129 of PRNP is considerably different between Europeans (32%-45%) and East Asians (92%-94%) normal populations (Table 3).
Codon 219 SNP
The PRNP codon 219 SNP introduces an amino acid substitution of lysine (Lys) for glutamic acid (Glu) (36). The SNP at codon 219 has been reported in Asian but not Caucasian populations (36, 37, 43, 55, 66). This SNP was linked to the development of sporadic CJD in the Korean and Japanese populations (36, 37).
Other PRNP polymorphisms
The deletion of the PRNP octapeptide repeat was not associated with sporadic CJD in the British population (67). Several SNPs outside the coding region of PRNP have also been investigated. The PRNP1368 polymorphism was associated with sporadic CJD in the British and German populations (68, 69). However, this finding could not be confirmed in the Korean population (70). Case-control studies in a Dutch population have shown contradictory results (71, 72). The PRNP -101, 310 and 385 SNPs showed a significant association with an increased risk of developing sporadic CJD after adjusting for the PRNP codon 129 genotype (73, 74, 75).
POLYMORPHISMS IN OTHER CANDIDATE GENES
Previous association studies of several genes other than PRNP have been performed in Europeans and East Asians (Table 4). For example, the prion-like protein gene (PRND), shadow of PrP (SPRN), cathepsin D (CTSD), HECTD2, tau protein gene (MAPT), apolipoprotein E (APOE), alpha1-antichymotrypsin (ACT), a disintegrin and metalloprotease 10 (ADAM10), ribosomal protein SA (RPSA), 14-3-3 eta (YWHAH), 14-3-3 beta (YWHAB), beta site APP cleaving enzyme 1 (BACE1), and calcium homeostasis modulator 1-3 (CALHM1-3) have all been investigated for relationships with human prion diseases.
PRND
PRND, the gene encoding the downstream prion-like protein (doppel or Dpl), is located downstream of human PRNP (76). Two SNPs in PRND, T26M and/or P56L, were not associated with sporadic CJD (68, 71, 77, 78). The T174M polymorphism has been inconsistently linked with sporadic CJD (68, 71, 77, 78, 79, 80). An association between sporadic CJD and a polymorphism in the 3' untranslated region (UTR) +28 position of PRND has been reported in the Korean population (81).
CTSD
CTSD, the gene encoding cathepsin D, is located on chromosome 11 (84). Cathepsin D co-localizes with PrPSc (85). CTSD C224T was associated with an increased risk of the development of variant CJD in the British population (86). However, this polymorphism was not associated with an increased risk of sporadic CJD in Korean or European populations (87, 88).
HECTD2
HECTD2, an E3 ubiquitin ligase, is located on chromosome 10. SNPs in HECTD2 have been associated with variant and sporadic CJD in the British population (89). However, the -247G>A and +16066T>A polymorphisms were not associated with genetic susceptibility to sporadic CJD in a Korean population (90).
RPSA
Ribosomal protein SA (RPSA), also known as 37 kDa laminin receptor precursor (LRP)/67 kDa laminin receptor (LR), is located on chromosome 3. LRP/LR acts as a receptor for PrPC and PrPSc (106, 107). Four RPSA SNPs (5' UTR-8T>C, 134-32C>T, 519G>A, and 793+58C>T) were not linked to sporadic CJD susceptibility (108).
CALHM1-3
The CALHM1 gene, which encodes a calcium homeostasis regulator, is located on chromosome 10 and controls cytosolic calcium levels (117). Two SNPs (rs41287502 and rs4918016) were associated with sporadic CJD in a Spanish population. However, two SNPs (rs2986016 and rs2986017) in CALHM1 and two SNPs (rs2986035 and rs3014199) in CALHM3 were not found to be associated with sporadic CJD in the Spanish population (118).
GENOME-WIDE ASSOCIATION STUDIES (GWAS)
Recent GWAS have been performed to identify genetic susceptibility factors for human prion diseases, including kuru, variant CJD and sporadic CJD (119). The rs6794719 and rs1460163 SNPs in the region upstream of RARB, which encodes retinoic acid receptor β and STMN2, which encodes the SCG10 protein, were strongly associated with kuru and variant CJD in a British population (38). These two SNPs in RARB and STMN2 were not associated with sporadic CJD in a Korean population (120). In addition, rs4921542, in the intronic region of the myotubularin-related protein 7 gene (MTMR7), and rs7565981, in the neuronal PAS (per-ARNT-sim) domain-containing protein 2 gene (NPAS2), were associated with variant CJD in the British and French populations but not sporadic CJD in three countries (39). Finally, SNPs at the ZBTB38-RASA2 locus were correlated with sporadic CJD in a British population but not a German population. A SNP in the CHN2 gene was associated with variant CJD but not sporadic CJD in a British population (40).
CONCLUSION
The human prion diseases are fatal neurodegenerative disorders characterized by the accumulation of PrPSc. Sporadic and genetic forms of human prion diseases are occurring worldwide. There is no doubt that mutations and polymorphisms in the PRNP play an important role in determining prion disease susceptibility. In this review, we have summarized that the proportion of PRNP mutations was quite significantly different between Europeans and East Asians and PRNP polymorphisms such as codons 129 and 219 were associated with sporadic CJD in the Europeans and East Asians. Nevertheless, the SNPs of other genes, including PRND, CTSD, HECTD2, and APOE have been showed contradictory results. Because GWAS studies have been reported in only the Europeans, these studies in the East Asians will be necessary to confirm and identify candidate genes for human prion diseases. In the future, the identification of new candidate gene in human prion diseases will contribute to understand numerous questions and potential therapeutic targets in prion diseases.



 XML Download
XML Download