

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Congenital pulmonary lymphangiectasia (CPL) is a rare primary developmental defect of the lung characterized by subpleural, interlobular, perivascular, and peribronchial lymphatic dilatation (1). The condition was originally described by Virchow (2) in 1856. Although the exact incidence of CPL is unknown, autopsy studies suggest that approximately 0.5%-1% of infants who are stillborn or die in the neonatal period have CPL (3). Patients with CPL usually present in the first few hours of life with intractable respiratory failure, cyanosis, and hypoxia associated with bilateral chylothorax. In an 18-yr retrospective study, Mettauer et al. (4) reported that 6 of 7 patients with pulmonary lymphangiectasia died. They concluded that although lymphangiectasia was not the direct cause of death in 2 of these patients, the prognosis of this disease was poor (4). Another study reported a mortality rate in cases of neonatal pulmonary lymphangiectasia of nearly 100% (5). However, recent advances in neonatal care have significantly improved the survival in cases of CPL with neonatal-onset (1, 6, 7).
In the present report, we describe the case of a newborn with CPL who survived after pneumonectomy of the affected lung.
CASE DESCRIPTION
A female infant was born at a gestational age of 39 weeks and 3 days by spontaneous vaginal delivery after an uncomplicated pregnancy. She was the second child of non-consanguineous parents. Prenatal examination, including ultrasonography, was normal. The infant weighed 3,650 g at birth. The Apgar scores were 9 and 10 at 1 and 5 min, respectively. Cyanosis and tachypnea were noted within 1 hr after birth, and the infant was transferred from a local clinic to our hospital on September 3, 2011. On admission, she required aggressive high-frequency oscillation ventilator support and surfactant replacement at an early stage in the course of treatment, which included antibiotics, inhaled nitric oxide, and catecholamines.
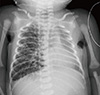
Chest radiography showed unilateral overinflation and interstitial thickening (Fig. 1). Moreover, the patient's respiratory symptoms worsened. On day 6, we performed high-resolution computed tomography, which is a standard diagnostic examination for congenital lobar emphysema (CLE) (Fig. 2). We noted that the hyperinflated right lung produced a mediastinal and cardiac shift to the left, compressing the left lung and aggravating the respiratory symptoms. We elected to completely resect the involved right lung in order to restore function of the contralateral lung. In pneumonectomy, double-lumen endotracheal tube was used for protection and separation of the contralateral lung.
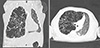
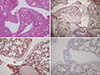
Histologic examination indicated the presence of CPL (Fig. 3 and 4). On gross pathological examination, the right lung was firm in consistency. The cut surfaces of the lung showed a network of numerous dilated cystic spaces ranging from approximately 1 to 5 mm in size (Fig. 3). Histologic examination of hematoxylin- eosin-stained tissue samples showed diffuse and marked cystic dilation of lymphatic vessels in the subpleural connective tissue, whereas the interlobular septa were widened and edematous (Fig. 3). No other specific pulmonary abnormalities, such as lymphatic proliferation or increased number of complex or anastomosing lymphatic channels, were noted. The dilated lymphatic cysts were lined with an attenuated simple endothelium, which was immunohistochemically positive for D2-40 and CD34, but was immunohistochemically negative for CD31 and pan-cytokeratin (Fig. 4). No evidence of CD68-positive foreign-body type histiocytes was noted.
Screening for congenital infection was negative, and the brain and abdominal ultrasounds were all normal. The karyotype was normal XX, and echocardiogram was normal.
During a follow-up examination at 24 months of age, the infant was well with weight and height above the 25th percentile, head circumference above the 50th percentile, and normal development.
DISCUSSION
CPL is a part of a spectrum of lymphatic disorders that are less well-characterized than other vascular tumors and malformations (8). CPL is usually bilateral but congenital unilobar pulmonary lymphangiectasia has been reported (9). The etiology of CPL is unclear, but one hypothesis suggests that the large lymphatic vessels of the lung that normally form at 9-16 weeks of gestation do not undergo normal regression at 20 weeks in these cases (10).
Noonan et al. (11) classified the patients with this condition into 3 groups. Patients in group 1 have generalized lymphangiectasia that may include hemihypertrophy, and pulmonary and intestinal involvement. Pulmonary involvement is less severe in this form and has a better prognosis. Patients in group 2 have acquired dilated pulmonary lymphatic channels secondary to the obstruction of pulmonary venous flow, such as that associated with total anomalous venous return. Group 3 represents patients who may have a primary developmental defect of the pulmonary lymphatic vessels that occurs after the 16th week of fetal life (12). CPL in Noonan's group 3 usually follows an adverse clinical course and is associated with high mortality (11). Faul et al. (13) recently proposed a new classification of these diseases, based on clinical presentation and pathologic features rather than the assumed pathophysiology. According to this classification, lymphangiectasia is categorized into primary (congenital) and secondary forms. The primary form has a neonatal onset and is fatal. The secondary form results from impaired lymphatic drainage and increased lymph production and develops during childhood. Faul et al. (13) proposed that primary and secondary lymphangiectasia could be distinguished by the age of the patients and their clinical courses. Our case is an example of pulmonary lymphangiectasia without associated cardiovascular abnormalities. It would be classified as group 3 by Noonan's classification (11), and as primary pulmonary lymphangiectasia according to the criteria of Faul et al. (13).
As observed in the present case, CPL tends to be misdiagnosed as CLE because of the clinical and radiographic similarities between the 2 entities (14). No clear clinical distinction exists between these diseases, and a definitive diagnosis of CPL can be made only by pathologic examination (3, 11, 14). CPL shows positive staining for D2-40, vimentin, CD31, CD34, and factor VIII-related antigen (15). The patient in the present case was immunohistochemically positive for D2-40 and CD34, which was consistent with a diagnosis of CPL.
Pulmonary interstitial emphysema (PIE) is a frequent complication of neonatal respiratory distress syndrome (16). PIE and CPL have clinical and pathologic similarties (16), such as massive dilatation of the interstitial lymphatic vessels. Both PIE and CPL also show D2-40-positive immunohistochemical staining of the inner lumens of the cystic lesions in the pulmonary parenchyma and the dilated lymphatic vessels (17). However, these diseases also have pathologic differences. In PIE, the giant cell reaction that forms a partial lining of the air spaces is a characteristic microscopic feature, indicating a reaction to the interstitial air (16, 17, 18). This finding was absent in our case.
CPL should be distinguished from diffuse pulmonary lymphangiomatosis (DPL) (8, 19), which cannot be differentiated immunohistochemically from CPL. DPL is characterized by an increased number of complex anastomosing lymphatic channels, with secondary variable dilation or expansion within the lungs and mediastinum. In contrast, the lymphatic vessels in CPL are normal in number and are relatively more regular in size and shape (8, 16).
Treatment is mostly supportive. Intubation and mechanical ventilation, drainage of pleural and peritoneal effusions, and correction of hypoxia, acidosis, and shock may be required in the delivery room to stabilize the infant's clinical condition. High-frequency oscillatory ventilation and/or nitric oxide and extracorporeal membrane oxygenation may be indicated for the treatment of persistent pulmonary hypertension (19). Surgical resection can be curative in selected cases (4, 5, 6). No cases of pneumonectomy for pulmonary lymphangiectasia have been previously reported in a newborn. Yalcin et al. (20) recently reported their experience of pneumonectomies of childhood involved a case of congenital pulmonary lymphangiectasia. But their patients were 8 yr old (0.5 to 17 yr) on a median age.
In the present case, pulmonary lymphangiectasia involved the entire right lung and we elected to perform right pneumonectomy to treat the severe respiratory distress. We plan to carefully follow-up the patient for the detection of several potential postpneumonectomy complications. Although the long-term outcome of CPL is uncertain, current evidence suggests that the symptoms gradually improve over time, particularly in cases where there are no significant coexisting abnormalities (1).

 XML Download
XML Download