

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Arterial restenosis is the major complication in vascular reconstructive procedures such as bypass graft, endarterectomy, balloon angioplasty and stent angioplasty. Restenosis is caused by intimal hyperplasia, a pathological process involving vascular inflammation, migration and proliferation of smooth muscle cells, and remodeling of extracellular matrix (1, 2). Therefore, the identification of novel targets for intervention of intimal hyperplasia is necessary to prevent postoperative restenosis (3-5).
Mechanical injury to the vessel wall triggers the release of a number of growth factors and cytokines, including platelet-derived growth factor, fibroblast growth factor, insulin-like growth factor, monocyte chemotactic protein-1, interleukin 6 and vascular endothelial growth factor, from endothelial cells and smooth muscle cells through activation of various signaling pathways, such as nuclear factor-kappaB (NFκB) (6, 7). These vascular responses lead to recruitment of inflammatory cells and the migration and proliferation of endothelial cells and smooth muscle cells. It has been thought that aberrant wound-healing responses to injury are involved in the development of intimal hyperplasia (8), but the mediators implicated in abnormal responses have yet to be fully identified. Therefore, a variety of gene knockout mice have been examined in an attempt to explore the molecular mechanisms using murine models of intimal hyperplasia, including carotid artery ligation.
Transglutaminase 2 (TG2 or tissue TG) is a calcium-dependent enzyme that catalyzes transamidation reaction between glutamyl residue of protein and either lysyl residue of protein or polyamines, producing crosslinked or polyaminated proteins (9). By these modifications of substrate proteins, TG2 is involved in apoptosis, differentiation, cell adhesion and extracellular matrix formation (10). Previously, we showed that TG2 is latent under unstimulated conditions, and activated by oxidative stress or tissue injury through transforming growth factor beta (TGFβ) signaling pathway (11). In addition, TG2 activated by tissue injury triggers inflammatory and fibrogenic responses by crosslinking IκB and fibronectin, respectively (12, 13).
TG2 is abundantly expressed in cardiomyocytes, endothelial cells, smooth muscle cells and macrophages. TG2 is reported to be involved in a number of cardiovascular diseases (14). Overexpression of TG2 in ventricular myocytes resulted in cardiac failure by upregulation of COX-2 expression (15). TG2 released from injured arteries is involved in the formation of arterial calcification through induction of chondro-osseous differentiation of smooth muscle cells (16). Recently, it has been reported that TG2 regulates the inward remodeling that occurs after reduction of blood flow in the rat mesenteric arteries (17). Inhibitors of TG2 prevented inward remodeling induced by endothelin-1. Conversely, expression of endogenous TG2 or the application of exogenous TG2 enhanced inward remodeling in several animal models (17). Moreover, inward remodeling induced by ligation of mesenteric artery was delayed in TG2-deficient mice (18).
These findings suggest that overstimulation or aberrant activity of TG2 may be implicated in the abnormal wound-healing responses. However, the role of TG2 in the process of intimal hyperplasia has not been elucidated yet. In this study, we compared the neointima formation induced by carotid artery ligation or loop injury between TG2-deficient and wild-type mice, and analyzed the expression and activity of TG2 in the carotid artery after arterial stenosis.
MATERIALS AND METHODS
Animals
Mice deficient for TG2 (TG2-/-) (19) and wild-type C57BL/6J strain were used. Wild-type mice were purchased from Japan SLC, Inc. (Hamamatsu, Japan) and bred for 4 weeks before experiments. TG2-/- mice were in-bred in the institutional research animal center (Seoul, Korea). Mice were fed ad libitum and had free access to drinking water. Male mice at 12 to 16 weeks of age were used for the experiments.
Mouse model of intimal hyperplasia
Mice were anesthetized by intraperitoneal injection of a mixture of zoletil (10 mg/kg, Virbac Korea, Inc., Seoul, Korea) and rompun (10 mg/kg, Bayer Korea Inc., Seoul, Korea). After a midline cervical incision, the left carotid artery was dissected. For ligation model, the common carotid artery just proximal to the bifurcation was ligated with 6-0 black silk (20). For loop injury model, the external carotid artery was opened and a prolene loop was inserted proximally up to proximal common carotid artery. Intimal injury was induced by twisting and traction of the loop, and then external carotid artery was ligated (21). Right carotid artery was used as an internal control. After 2 or 4 weeks, both carotid arteries were harvested. For intracellular TG activity assay, fresh tissues were transferred to culture media. For Western blotting, tissues were snap-frozen in liquid nitrogen.
Histochemistry and quantitative morphometry
Arteries were fixed in situ by perfusion with saline and 4% paraformaldehyde, then embedded in paraffin. Thin sections (5 µm) were stained with hematoxylin & eosin (H&E). Using an Image J program 1.42q (National Institutes of Health, Bethesda, MD, USA), the areas of lumen, intima and media of carotid artery were measured. Numbers of stained cell in the intima were counted. All data were obtained in a blinded fashion at a magnification of *400. Masson's trichrome staining was performed to compare the collagen contents. Cell proliferation and apoptosis were assessed by immunohistochemical staining of proliferation cell nuclear antigen (PCNA) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, respectively. TG2 expression was examined by immunohistochemical staining using monoclonal antibody specific for TG2 (22).
Intracellular transglutaminase (TG) activity assay
In situ TG activity in the arterial tissue was analyzed by Western blotting and microtiter plate assay as described elsewhere (23). In brief, the sliced arteries (carotid artery and descending thoracic aorta) were incubated with serum-free DMEM containing 3 mM biotinylated pentylamine (BP; Pierce, Rockford, IL, USA) for 1 hr at 37℃, 5% CO2 in 24 well culture plates. The arteries were grinded by glass beads in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Trixton X-100 and protease inhibitor cocktail, Roche Korea Co., Seoul, Korea). The homogenates were centrifuged at 12,000 g, 4℃ for 10 min. The protein concentration of supernatants was determined by the bicinchoninic acid assay method. For Western blot analysis, the cell extracts were subjected to SDS-PAGE using 10% gel and then transferred to nitrocellulose membrane. The proteins that crosslink with BP were visualized by probing with horseradish peroxidase (HRP)-conjugated streptavidin (Zymed laboratory Inc., South San Francisco, CA, USA), followed by enhanced chemiluminescence reagents (Pierce). The bands intensity was measured using Image J. For microtiter plate assay, the cell extracts (5-10 µg/well) were added to each well of a 96-well microtiter plate (Nunc) and coated for 1 hr at 37℃. After incubating HRP-conjugated streptavidin for 1 hr at room temperature, TG activity was quantified by the reaction with O-phenylenediamine dihydrochloride (Sigma-Aldrich Korea, Yongin, Korea) and measuring absorbance at 492 nm on microplate spectrophotometer (Molecular Devices) after the reaction was stopped by adding 1 M H2SO4.
Statistics
All values are expressed as mean±standard deviation. Statistical analysis was conducted via unpaired Student's t-test and Mann-Whitney test using SPSS software (version 17.0, SPSS Inc., Chicago, IL, USA) or one-way ANOVA with test for linear trend using GraphPad Prism (version 5.00 for Windows, Graphpad Software, San Diego CA, USA). P values of <0.05 were considered statistically significant.
RESULTS
Intimal hyperplasia in loop-injured carotid arteries and in ligated arteries in TG2-null mice
TG2 is implicated in apoptosis, cell adhesion and extracellular matrix formation under stressed conditions. To define the role of TG2 in the process of intimal hyperplasia, we examined the effect of TG2 gene ablation on neointima formation by using an in vivo model. The common carotid artery of wild-type and TG2-/- mice were ligated with black silk (n=8). The ligated and control carotid arteries were harvested 2 and 4 weeks after operation, and H&E staining of carotid artery was performed. Morphometric analysis of H&E sections under light microscopy (×400) showed that no intimal hyperplasia or luminal narrowing of ligated arteries was developed in the carotid artery of wild-type mice after 2 and 4 weeks when compared to control. Moreover, there was no significant difference in the development of intimal hyperplasia of carotid artery between wild-type and TG2-/- mice after ligation (Fig. 1A, B). These results indicate that hemodynamic stimuli induced by ligation are not associated with the development of intimal hyperplasia in carotid artery, suggesting that artery ligation is not appropriate model to elucidate the role of TG2 in the neointima formation.
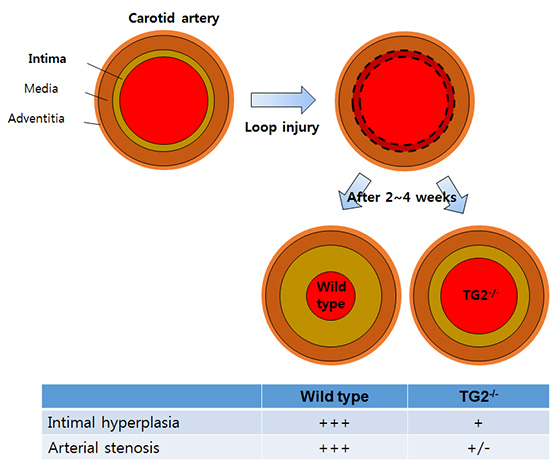
Next, we tested whether TG2 is involved in the intimal hyperplasia in response to tissue damage. The external carotid artery of wild-type and TG2-/- mice was opened, and intimal injury was induced by twisting and traction of the prolene loop after inserting up to proximal common carotid artery. In both wild-type and TG2-/- mice, H&E staining of carotid artery showed a gradual increase in intimal thickness in response to loop injury after 2 and 4 weeks (Fig. 2A, B). Morphometric analysis revealed that an area of intima measured at 4 weeks was about 2.5 folds larger than that measured at 2 weeks after operation (n=20 for each group; Fig. 2C). When compared the arterial structure between wild-type and TG2-/- mice, the area of intima was markedly reduced in TG2-/- mice both after 2 and 4 weeks, although there was no statistical significance (P=0.063 after 2 weeks, P=0.218 after 4 weeks; Fig. 2C). By contrast, the area of media was similar in carotid arteries from wild-type and TG2-/- mice, both after 2 and 4 weeks (Fig. 2D). Therefore, the relative intimal thickness, expressed as a ratio of intimal/medial area, was significantly reduced in carotid artery of TG2-/- mice (P=0.007 at 2 weeks, P=0.043 at 4 weeks; Fig. 2E). These results indicate that TG2 is involved in the development of intimal hyperplasia, but not medial changes, in response to vascular injury.
Expression of TG2 after vascular injury
To gain insight into the mechanism by which TG2 play a role in intimal thickening, we first examined the expression of TG2 in carotid artery after vascular injury. Immunohistochemical staining with monoclonal antibody specific for TG2 revealed that intimal cells of carotid artery were densely stained at 2 weeks after vascular injury. After 4 weeks, immunoreactive cells, which were diffusely stained, appeared in media as well as in intima of carotid artery, but no vascular cells in the adventitia were stained (Fig. 3). These results indicate that expression of TG2 is upregulated by vascular injury.
Previous reports showed that intracellular TG2 is latent in various epithelial cell lines, and activated under oxidatively stressed conditions, such as ischemia-reperfusion and hypoxia, through increase of intracellular calcium level or TGFβ signaling pathway (11, 24). To test whether TG2 could be activated by vascular injury, we measured the intracellular activity of TG2 in the arterial tissues. Carotid arteries and descending thoracic aorta were harvested at 2, 4, and 5 hr after loop-injury from wild-type and TG2-/- mice, and were incubated in culture medium containing biotinylated pentylamine (BP). TG activity in vascular tissues from wild-type mice, determined by measuring the amount of BP-incorporated proteins with Western blotting, was increased about 2-2.5 folds and peaked at 4 hr after loop-injury (Fig. 4A, B). Notably, a substantial increase of TG activity was observed in the vascular tissues from TG2-/- mice, suggesting the expression of other type of transglutaminase family, probably coagulation factor XIII (18). Moreover, when compared the intracellular TG activity between the loop-injured and non-injured carotid arteries harvested at 4, 7, 14, 28 days after surgery, the TG activity was gradually increased throughout a time period of 4 weeks in injured arteries, whereas the TG activity remained in relatively low levels during 4-week periods in non-injured arteries (Fig. 4C).
Mechanical injury to intima provokes inflammatory responses that induce the proliferation and migration of vascular smooth muscle cell (VSMC) (1, 5). To test whether TG2 is involved in the proliferation of VSMC, we examined the expression of PCNA in carotid arteries after 2 and 4 weeks of intimal injury. Immunohistochemical analyses of carotid arteries from wild-type and TG2-/- mice showed no difference in the number of PCNA-positive cells between two groups (data not shown). Moreover, since TG2 plays an anti-apoptotic role under oxidatively stressed conditions, we evaluated the apoptosis of VSMC in injured-carotid artery. TUNEL staining revealed no difference in the number of apoptotic VSMC between wild-type and TG2-/- mice at both time points (data not shown). Considering the recent report showing the critical role of TG2 in the development of fibrosis (12), these results suggest that TG2 might be responsible for the formation of extracellular matrix in the late response to intimal injury.
DISCUSSION
Arterial restenosis occurs in a significant number of patients treated with various open and endovascular approaches for atherosclerotic stenosis of coronary, cerebrovascular and peripheral arteries, limiting the success of surgical interventions. Therefore, the identification of novel targets to prevent the postoperative restenosis is an important research topic (3, 4). In the present study, we showed that intimal thickness of carotid artery after endothelial denudation is reduced in TG2-deficient mice compared to that of wild-type mice. Our data indicate that in a mechanically-induced endothelial denudation model, TG2 activity is responsible for the development of intimal hyperplasia.
The injury to vascular wall triggers wound-healing responses, comprising migration of smooth muscle cells from the media to the intima, subsequent proliferation of intimal cells, and deposition of extracellular matrix to form a neointima (3, 5). As demonstrated by several mouse models, arterial narrowing after vascular injury is caused by inward remodeling of artery or intimal hyperplasia, an exaggerated wound-healing response (1, 2). In this study, the two most commonly used procedures, carotid artery ligation and mechanical denudation of the endothelial layer, were employed to induce vascular injury. In a carotid artery ligation model, our data showed that there is no significant difference in neointimal formation between wild-type and TG2-deficient mice after 2 or 4 weeks of ligation. By contrast, previous study using a small mesenteric artery ligation model demonstrated that TG2 and macrophage-derived coagulation factor XIII, other isoenzyme of transglutaminase family, are involved in the process of inward remodeling (18).
In contrast to the results of ligation model, we found that intimal hyperplasia was significantly reduced in carotid artery from TG2-deficient mice after endothelial denudation. Moreover, TG2 expression is increased in neointimal and medial cells after intimal injury, and intracellular TG2 in large artery is activated by endothelial denudation. Since the endothelial denudation model resembles the clinical setting of vascular intervention, our results suggest that TG2 plays a causal role in the development of postoperative restenosis. In addition, because hemodynamic parameters are maintained in this model, changes in shear stress or endothelial dysfunction may be not contributing factors to the development of denudation-induced intimal hyperplasia (25). Therefore, comparison of vascular response between two mouse models indicates that the mechanism responsible for intimal hyperplasia in the endothelial denudation model may be different from that in the ligation model.
TG2 mediates the post-translational modification of a variety of proteins by catalyzing the transamidation reactions, producing cross-linked, polyaminated or deamidated proteins (26). Previously, we showed that TG2 induces protein aggregation by polyamination of proteins (11), inhibits apoptosis by crosslinking of caspase 3 (27), and promotes inflammation by activation of NFκB signaling pathway (13). Therefore, an important role for aberrant activation of TG2 has been proposed in the pathogenesis of neurodegenerative diseases, celiac disease, cataract formation, and fibrosis (28). In fact, TG2 is unique among 9 member of TG family (TG 1-7, coagulation factor XIIIa, band 4.2) in that TG2 is activated by various types of oxidative stress, such as UV-radiation, hypoxic stress and bleomycin treatment. Although the exact mechanism that regulates the activation of intracellular TG2 is not fully understood, oxidative stress activates TG2 through calcium or TGFβ signaling pathway (11). As shown in this study, mechanical injury upregulates the TG2 expression, and also activates TG2. These data suggest that TGFβ released by mechanical injury may activate TG2 which subsequently activates NFκB signaling pathway. Thus, it is a possible speculation that a number of cytokines produced by activation of NFκB may be implicated in the development of intimal hyperplasia after endothelial injury.
Currently, angioplasty and stenting have become routine procedures for the treatment of vascular occlusive diseases. One of the major complications limiting the success of these procedures is intimal hyperplasia, which leads to restenosis in a substantial proportion of patients (4). Therefore, understanding the cellular and molecular mechanisms for the development of intimal hyperplasia is an important issue to identify the novel target for prevention or treatment of intimal hyperplasia after vascular surgery. Our data show that intimal injury increases the TG2 expression and enzyme activity that contributes to the development of intimal hyperplasia. Although some drug eluting stents in coronary artery stenosis have been shown to reduce the risk of restenosis, at present, there is no systemic pharmacological intervention to prevent the intimal hyperplasia (4). Therefore, the mechanism based inhibitors that we have investigated might provide a basis to develop new compounds that could be used either to prevent or delay the progression of restenosis.
In summary, the purpose of this study is to define the role of TG2 in the process of intimal hyperplasia and inward remodeling using animal models. In an endothelial denudation model, we show that the development of the neointima was significantly reduced in TG2-null mice, suggesting that TG2 plays a critical role in the development of intimal hyperplasia after vascular injury.




 XML Download
XML Download