

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Bordetella pertussis is the pathogenic organism responsible for pertussis. Infants younger than 6 months old are a high-risk group (1). As vaccination can prevent pertussis, the global incidence of reported pertussis cases was dramatically reduced after the introduction of vaccination. However, continuous pertussis outbreaks in countries where vaccination is standard have given rise to major concerns about the control of the disease (2). Various studies suggested that the waning of vaccine immunity (3-5) and the emergence of genotype variants are responsible for the outbreaks (6-8). To improve herd immunity in adolescents and adults, an adult pertussis vaccine was introduced, and vaccination was recommended (9, 10). Genetic changes observed in the strains were mostly concentrated in antigenic determinant genes, which encode vaccine components, such as pertussis toxin, filamentous hemagglutinin, pertactin, and fimbriae (11, 12). More recently, another variant type was also reported (12). This variant was inferred as a more toxic strain because the production of pertussis toxin was increased 2-3 times by a point mutation in the promoter region (13).
In Korea, the number of reported cases of pertussis started to increase from 2009 onward. As shown in Fig. 1, the number of reported pertussis cases was relatively constant during 2000-2008. At this stage, average number of reported pertussis cases was 11.5 cases/yr. However, the reported number of cases rapidly increased to 66 in 2009. At that time, we analyzed the genotype variations to confirm whether there were genotype changes in the strains and we reported the emergence of ST2 strains, which accounted for 58.3% of the strains (14). After the publication of that report, the numbers of pertussis cases increased to 97 cases in 2011 and to 230 cases in 2012. Therefore, we further analyzed genetic variations in 21 strains isolated in 2011-2012 by multilocus sequence typing method. In the present study, we present the results of the comparison of the strains isolated in 2009 with those isolated in 2011-2012.
MATERIALS AND METHODS
Bacterial isolates and culture conditions
Twenty-one B. pertussis strains isolated between 2011 and 2012 in Korea were analyzed (Fig. 1). These clinical strains were directly isolated from nasopharyngeal swab or sputum. These specimens were received from hospitals as part of public diagnostic services of pertussis or from affiliated hospitals as part of a pertussis surveillance network. These specimens were directly inoculated on Reagan Lowe medium (10% horse blood) with cephalexin and incubated in a humid incubator at 37℃ for 7 days. After incubation, a suspected colony was confirmed by the slide agglutination test with Bordetella antisera (Difco), and also confirmed by PCR with species-specific primer sets (BP and pTp) as described in our previous report (14). All the isolated strains were preserved in a deep freezer (-80℃) until analyzed. In addition to these clinical isolates, 3 reference strains (ATCC 10380, ATCC 9797, and Tohama I) were included. The genomic DNA of the isolates was prepared with a commercial kit (GenEx genomic Sx kit, GenaAll Biotechnology, Seoul, Korea) according to the manufacturer's instruction.
Genotype analysis of target genes
The target genes for MLST analysis were 7 housekeeping genes (adenylate kinase, fumarate hydratase class II, aromatic amino-acid aminotransferase, isocitrate dehydrogenase, cytosol aminopeptidase, and phosphoglucomutase) and 10 antigenic determinant genes (pertussis toxin subunit 1, pertactin, filamentous hemagglutinin, fimbriae2, fimbriae3, outer-membrane protein q, Bordetella antitransporter-protein c, adenylate cyclase toxin, virulence-activated gene, and tracheal colonizing factor). The PCR primers and PCR conditions of most of the test genes were those cited in our previous report (14) and in the Bordetella multilocus sequence typing web site (http://pubmlst.org/bordetella/). After amplification of the target genes, their nucleotide sequences were confirmed by sequencing.
The genotype determination of the target genes was performed as described in our previous report (14). Briefly, for the housekeeping genes, the sequence information of the tested strains were queried using the Bordetella MLST database. For the antigenic determinant genes, the sequences of the tested strains were compared with the DNA sequences of each reported genotype in the NCBI GenBank by the MEGA program (15).
Multilocus sequence type analysis
For the sequence types of the housekeeping genes, the genotype profile of each isolate was queried using the Bordetella MLST database. For the antigenic determinant genes, we independently assigned sequence types (STs) according to the frequencies of genotype profiles as described in our previous report (14). The relatedness among each ST was analyzed by the START program package (16). The evolutionary relationship and clonal complexes (CCs) among the STs were analyzed and represented as a minimum spanning tree using the PHYLOViZ program (17).
Analysis of pertussis toxin promoter variations
To confirm the sequence variations in the pertussis toxin promoter region, a 550 bp target region was amplified using a specific PCR primer set (forward: 5'-AATCGTCCTGCTCAACCGCC-3', reverse: 5'-GGTATACGGTGGCGGGAGGA-3') (13). After the PCR, the amplified PCR products were sequenced. The confirmed nucleotide sequences were compared to the reference sequences listed in GenBank (13) using alignment.
RESULTS
Genotype profiles of housekeeping genes
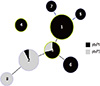
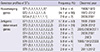
According to the Bordetella MLST database, 43 STs and 4 CCs were observed in the Bordetella genus (3 species, B. pertussis, B. parapertussis, and B. bronchiseptica) (18). Of these 4 CCs, CC2 belongs to the B. pertussis species and is composed of 3 sequence types, ST1, ST2, and ST24. Three similar genotype profiles of 7 housekeeping genes containing these 3 STs (ST1, ST2, and ST24) were observed in this study by querying the Bordetella MLST database. Among these STs, the most high frequent was ST2 (Table 1), and it was continuously recorded since 1999. However, frequencies of the other 2 STs (ST1 and ST24) were very low (Table 1) and these 2 STs observed in only old strains. As shown in Fig. 2, ST2 was confirmed as a major ST in the isolates from 2011-2012, with all the tested strains isolated in this period showing this ST.
Genotype profiles of antigenic determinant genes
In the analysis of the antigenic determinant genes, 8 STs were confirmed in the Korean isolates (Table 1). For this, the new genotype results confirmed in the isolates of 2011 and 2012 were combined with our previous results (14). The most frequent form was ST1 (57.8%), but this ST was not observed in isolates of 2010. The next frequent type was ST2 (23.5%), which was recognized 2009, was observed until 2012. As shown in Fig. 2, the STs confirmed in the isolates of 2011-2012 were ST2, ST3, and ST8. As reported earlier, ST8 first emerged in this period (14). In addition, the frequency of ST2 increased from 8.6% to 81.0% (P<0.001). The increase was attributed to an increase in ST2 in the isolates of 2011-2012.
Lineage assignment analysis of antigenic determinant genes
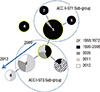
Compared to the results of the analysis of the housekeeping genes, the antigenic determinant gene group showed a more divergent tendency. Only 3 STs were confirmed in the housekeeping gene analysis, whereas 8 STs were confirmed in the analysis of the antigenic determinant genes (Table 1). In addition, although a single CC was confirmed in the antigenic determinant gene analysis, there appeared to be two subgroups, which originated from two central STs. In our previous report (14), we assigned these two subgroups to ACC I and ACC II. As depicted in Fig. 3, all the STs were grouped as a single CC, except ST4 (singleton). However, this single CC was divided into two subgroups, and these two subgroups were linked by two central STs such as ST1 and ST3.
Characteristics of the ST3 subgroup
As shown in Fig. 4, ST3 expanded and ST2 diverged from ST3 in 2009. From 2011, ST2 expanded, and ST8 diverged from ST2. In particular, the frequency of ST2 increased substantially. In this dynamic period of change in the ST of the circulating strains, reported numbers of pertussis cases also increased considerably (Fig. 1). To shed light on the relationship between the ST2 strain and the increase in pertussis cases, we further characterized the strains isolated in this period about promoter variation. As described, there were reports (12, 13) of more toxic strains showing increased pertussis toxin production due to a mutation in the promoter region (ptxP3 type). As expected, the ptxP3 type was first detected in 2009. The frequency of ptxP3 gradually increased from 53.8% in 2009 and reached 100% by 2012. As depicted in Fig. 5, the ptxP3 type was observed in only ST3, ST2, and ST8. Other STs showed the ptxP1 type. The emergence of the ptxP3 type was dependent on the isolation year because only the strains isolated later than 2009 showed the ptxP3 type, even in the same STs. In the case of ST3, the strains in ST3 were divided into three groups according to isolation year (2000, 2009, 2011). From these groups, only the strains isolated in 2011 showed ptxP3 type.
DISCUSSION
A dynamic change of antigenic genotypes in the pertussis pathogen was confirmed in this study. The change commenced in 2009 and continued until 2012. We have to consider two points: the change in the ST of the strains and the emergence of toxic strains showing the ptxP3 promoter type.
Genotype changes in the strains have already been reported in several other countries. Similar to our results, all the reported genotype changes were concentrated on antigenic determinant genes. Although genotype changes in housekeeping genes were also analyzed in this study, no changes were observed. In the Korean isolates, ST2 was the major genotype in the housekeeping genes since 1999 (Table 1). This result shows that there was selective pressure that affected only on the antigenic determinant genes. Currently, it is inferred as the vaccination effect (19). It is also supported by the trends of genotype changes of the strains toward a non-vaccine type. The representative genes reported in the present study were pertussis toxin, pertactin, fimbriae, and filamentous hemagglutinin. These proteins were the vaccine components of currently using acellular vaccine.
Similar results were also recognized earlier in Korea (14). Although Tohama I of the gene sets (ptxS1, Prn, FHA, fimbriae2, and fimbriae3) showed a 2,1,2,1,1 genotype profile, the genotype profiles observed in the current strains were 1,1,1,1,2 (ST1), 1,2,1,1,1 (ST2), and 1,2,1,1,3 (ST8). In addition, as shown in Fig. 4, the direction of the ST changes observed in the current strains was ST3 → ST2 → ST8. In this direction, the responsible genes for changing STs were prn and fim3. The prn gene is a 69 kDa outer membrane gene involved in adherence to host cells. The genotype was determined by the tandem repeat unit of amino acids (GGXXP) (20). Since 2009, the prn genotype of the circulating strains in Korea changed from type 1 (vaccine type) to type 2. In the case of the fim3 gene encoding fimbriae, type 3 emerged since 2011. This fim3 type 3 is a variant type of an amino acid change in the epitope region of fimbriae (19). The results imply that the recently confirmed genotype changes are due to changes in outer proteins related to immunogenicity. With respect to promoter type variation (13), the ptxP3 variant emerged since 2009 in ST2, ST3, and ST8. The proportion of the ptxP3 type in the 2009 isolates was 58.3%, and this increased to 86.6% in the 2011 isolates. Finally, all isolates of 2012 showed the ptxP3 type. Therefore, we conclude that the recently observed increase in pertussis cases in Korea is closely related to the emergence of variant strains.
Global trends reported for genetic variations were similar to our results, except for fimbriae (19). The genotypes of ptxS1, prn, and ptxP3 were similar to global trends. However, the fimbriae genotypes were different. The major genotypes of fim2 and fim3 according to global trends changed from fim2-1 to fim2-2 and from fim3-1 to fim3-2. In Korea, the fim2 genotype did not change (fim2-1), but the fim3 genotype changed from fim3-2 to fim3-1. These results indicate that the Korean strains are changing from the vaccine type in common with the global strain.
The acquisition mechanism of these variations is not clearly resolved. However, it was inferred that the insertion sequence element (IS481) has an important role in the adaptation of B. pertussis to its host (19, 21). The genetic change mediated by IS481is a large-scale chromosomal rearrangement. The genome reduction and gene loss are caused by this genetic event. According to one report, the genome size of current strains has been gradually decreasing since 1950 (21). Other genetic changes that have been recorded are In/Del polymorphisms in repeat units and single nucleotide polymorphisms (SNPs). The representative case for In/Del polymorphism was observed in the pertactin structural gene (20, 21). As described above, the variants in the prn gene are the result of changes in the number of short tandem repeat units and it was caused by homologous recombination. Another form of genetic changes frequently observed in the B. pertussis genome is SNPs (silent and non-silent SNPs). Especially, non-silent SNPs found in surface antigens cause structural changes in the epitope region and may affect the immune response (19, 21). In addition, SNPs affect regulatory elements of genes related to virulence. The representative case was the promoter variants of the pertussis toxin gene. Currently, 14 variants types are known and the variation was observed in the BvgA binding site, the global regulator of virulence gene expression. Among these variants, the ptxP3 type shows high expression (more than 2-fold) of the pertussis toxin (13) and has an effect on colonization in respiratory tract (22).
As noted above, there have been many reports of emerging genetic variants of B. pertussis strains since 1990. The remaining question from these reports is that the observed genotype polymorphisms were attributed in simple drift event or evolutional event (6). Currently, it might be concluded as evolutional event by active adaptation of the pathogen to avoid massive vaccination effects (19). Therefore, it is more important to predict the direction of genotype changes in current strains to cope with emerging new toxic variants. For this reason, the study of genotype variation is gradually moving toward comparative genomic studies to obtain extensive data occurred in whole genome (21). Although MLST analysis for bacterial typing allows rapid generation of clear results and easy comparisons with results (23), the data obtained from MLST analysis are restricted to only the selected gene group and are more suitable for a retrospective study. In addition, the discriminating power is less than that of other typing methods, such as pulsed-field gel electrophoresis (PFGE) or multiple locus variable-number tandem repeat analysis (MLVA) (24). PFGE is known as the gold standard for bacterial typing but it is too laborious and difficult to standardize (25). Therefore, PCR-based typing methods, such as MLVA and MLST, are used more frequently because these methods are relatively simple and easy. In addition, the discriminating power can be increased PFGE level, if MLVA data are combined with MLST data (25). Taken the above into consideration, the genotype variations of the Korean strains confirmed in this study are insufficient to predict future trends in emerging of toxic strain. Therefore, a comparative genomic study is needed between isolated Korean strains.
In conclusion, the present study confirmed that the change in the strains in Korea is an ongoing event. It cannot be predicted whether the ST2 strain will continue to be a major ST or whether other ST strains carrying the ptxP3gene will emerge. Therefore, the current surveillance system for pertussis should be maintained and genetic characterization of the strains according to their STs should be extended to the whole genome sequence level.



 XML Download
XML Download