

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hunter syndrome (or mucopolysaccharidosis type II [MPS II], OMIM# 309900) is a rare and life-limiting X-linked recessive disorder that affects approximately 1 in 77,000-94,000 newborn boys (1-3). The cause of the disease is a deficiency in the lysosomal enzyme iduronate-2-sulfatase (IDS, EC 3.1.6.13), which is responsible for breaking down glycosaminoglycans (GAGs) within cells (4). Impaired GAG degradation results in progressive damage of various tissues and organs (4). Affected individuals present with coarse facial features, short stature, skeletal deformities, and joint stiffness, and frequently show mental retardation (4, 5).
Short stature is a prominent and consistent feature of Hunter syndrome (5-7); this generally becomes evident when the individual is approximately 8-10 yr of age (5, 7, 8). During childhood, short stature may have a negative impact on their quality of life and social functioning (9-11). In addition, the physical growth of patients is of paramount importance in pediatric practice and is a highly emotive issue for parents. Thus, ameliorating short stature may be beneficial to patients and their parents.
Enzyme replacement therapy (ERT) with idursulfase (Elaprase®; Shire HGT Pharmaceuticals, Cambridge, MA, USA) has been available for patients with MPS II since August 2006 in the US, since March 2007 in Europe, and since February 2009 in Korea. More recently, ERT with idursulfase beta (Hunterase®; Green Cross Corp., Yongin, Korea) has been commercially available in Korea. A phase I/II clinical trial showed that idursulfase beta treatment was well tolerated in Korean patients with MPS II and resulted in a significant reduction in urinary excretion of GAGs and an improvement in the 6-min walking test distance (12). The effectiveness of ERT in pulmonary function and joint mobility was evaluated in several studies (13, 14).
To date, few studies have investigated the effect of ERT on the growth of individuals with MPS II (6, 15). No data are available for Asian MPS II patients, or for patients younger than 6 yr of age. The present retrospective analysis assessed the effect of ERT on the linear growth of Korean Hunter syndrome patients of different ages and phenotypes.
MATERIALS AND METHODS
Patient population and data collection
All the participants were recruited from the Department of Pediatrics in Samsung Medical Center. The diagnosis of MPS II was based on the clinical findings, as well as abnormal excretion of urinary GAGs and deficiency of IDS enzyme activity in skin fibroblasts or peripheral blood leukocytes. The disease was confirmed in all the patients by molecular analysis. Mutations of patient 4-32 were described previously (16). Those with cardiac failure, a history of hematopoietic stem cell transplantation or orthopedic surgery, and those on nocturnal respiratory support were excluded from this study. Patients on long-term oral steroid treatment and those who had previously received growth hormone therapy were excluded. Those who experienced episodes of anaphylaxis during ERT (17) and female patients with Hunter syndrome were also excluded. Patients who had commenced ERT (Elaprase® or Hunterase®) before they were 20 yr and had received ERT for at least 2 yr were included. All patients had received ERT (0.5 mg/kg) on a weekly basis, shortly after they were diagnosed or since 2009, when ERT became available in Korea.
The impact of ERT on growth was analyzed in patients for whom height data had been obtained on at least one occasion within 24 months before or after the start of the treatment. The height data were analyzed within 3 age-stratified groups. Two age cut-off points at the start of the ERT-6 yr and 10 yr-were selected because patients with Hunter syndrome tend to grow faster in their early years, followed by markedly reduced growth after around 8 yr of age onward. The patient's height was measured using a stadiometer (accuracy to 1 mm) according to standard protocols.
The clinical phenotypes of the patients were classified as severe if the patients had cognitive impairment, and as attenuated if they had no cognitive impairment even with pronounced bone and visceral involvement. Our previous study suggested that mild phenotype may be related to residual lysosomal enzyme activity (18). Type of mutation was determined based on the experience of the authors: mutations predicted to be associated with a severe phenotype (large deletion, recombination, deletion or nonsense mutation) and those predicted to be associated with a less-severe phenotype (splicing, insertion, insertion/deletion or missense mutation).
Data analysis
Three groups of patients were analyzed according to their ages at the start of the ERT (baseline): group 1 included patients under 6 yr of age (n=14), group 2 included patients aged 6-10 yr (n=11), and group 3 included patients aged 10-20 yr (n=7). The raw height data of the three groups were plotted against normal height data for boys from Korea. Height was expressed as the standard deviation score (SDS) based on normative data from Korean references. A z-score <-2 is generally considered to be indicative of short stature, and this score was used to plot the heights in the current study. For group 2 and group 3 with attenuated MPS II, the mean changes in the patients' heights and z-scores (±SD) from baseline were calculated.
A piecewise regression using a mixed model was used to analyze the age-corrected standardized height scores (z-scores) before and after the start of the ERT (±24 months of the start of the treatment) for the patients in groups 2 and 3. Individual analyses of the patients in groups 2 and 3 were conducted to assess covariates that were likely to impact on growth: age at start of ERT (6-10 vs 10-20 yr), type of mutation, and phenotype.
RESULTS
Patients
The demographic and molecular data of the 32 MPS II patients are listed in Table 1. The 32 boys started to receive ERT between the ages of 4 months until they were 19 yr 4 months (median age of starting ERT, 7 yr). All the patients were born at term and presented with typical clinical features of MPS II (median age of diagnosis, 3 yr 4 months). Cognitive involvement was evident in 13 of the 32 patients.
Effect of ERT on growth
In group 1 (n=14; age range at starting ERT, 4 months-5 yr 10 months), the heights of the patients at the initiation of ERT ranged from 78 to 119.4 cm (median, 106.3 cm). None of the boys were of short stature (<-2 SDS) at the start of the ERT, and the mean height SDS of the patients in group 1 was 0.48±1.71. The mean increase in height over the 2-yr period of ERT was 11.5±5.8 cm. The growth charts and the z-scores are shown in Fig. 1A and D. The growth curves during the ERT show that the height of 12 of these patients was still within the normal range based on normative data from Korean references. Two patients (patient 8, patient 13) whose height decreased to less than -2 SDS exhibited severe cognitive impairment when they were diagnosed with the disease. Changes in the z-scores and the growth velocities of these patients are not presented as figures because of a lack of data on the heights of the subjects prior to undergoing ERT.
In group 2 (n=11; age range at starting ERT, 6 yr 1 month-10 yr), the patients' heights at the initiation of ERT ranged from 98 to 122.5 cm (median, 116.2 cm). Six patients were considered to be of short stature at the start of ERT, and the mean height SDS of the patients in group 2 was -2.6±1.79. During 3 yr of ERT, the mean increase in height was 9.4±6.1 cm. The growth charts and the z-scores are shown in Fig. 1B and E. The yearly growth velocity was calculated for the seven patients in group 2 with attenuated MPS II. The increase in height was 3.1 cm (z-score=-0.22; one patient was excluded from this analysis due to lack of data of height before ERT) in the year before the ERT, compared with 4.9 cm (z-score=0.24), 3.8 cm (z-score=-0.02), and 4.2 cm (z-score=-0.04) in the first, second, and third years of the ERT, respectively (Fig. 2A, C).
In group 3 (n=7; age range at starting ERT, 10 yr 3 months-19 yr 4 months), the heights of the patients at the initiation of ERT ranged from 116.7 to 142.4 cm (median, 127.7 cm). Five patients were considered to be of short stature at the start of ERT, and the mean height SDS of the patients in group 3 was -5.12±3.79. During 3 yr of ERT, the mean increase in the height of the patients was 9.5±7.6 cm. The growth charts and the z-scores are shown in Fig. 1C and F. The increase in the heights of six patients in group 3 with attenuated MPS II was 0.7 cm (z-score=-0.47) in the year before the ERT compared with 3.9 cm (z-score=0.02), 2.5 cm (z-score=-0.01), and 3.6 cm (z-score=0.1) in the first, second, and third years of the ERT, respectively (Fig. 2B, D).
Analysis of the height z-scores of the 17 patients in groups 2 and 3
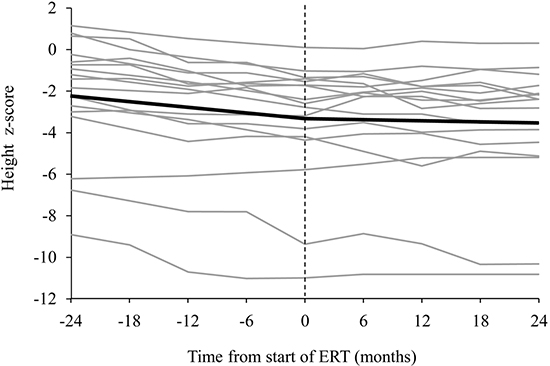
The overall analysis of the raw data using the height z-scores of the 17 patients who were aged 6-20 yr at the start of the ERT showed that the slope of the regression over time after the treatment (in months) was significantly changed compared with before the treatment (the estimated slopes before and after the treatment were -0.047 and -0.007, respectively: difference in the slope, 0.04; P<0.001) (Fig. 3A). The slope of the regression was particularly more changed in the 12 patients in groups 2 and 3 with the attenuated form of the disease (from -0.045 to 0.004: difference in the slope, 0.049; P<0.001). In contrast, the slope of the regression was not significantly changed in the 6 patients in groups 2 and 3 with the severe form of the disease (from -0.053 to -0.035: difference in the slope, 0.018; P=0.129).
Impact of age at start of ERT
Patients with Hunter syndrome aged 6-20 yr were shorter on average when compared to their peers. The height deficit was more pronounced in patients aged 10-20 yr of age at the start of treatment (difference in z-score at the start of ERT, 2.89; P=0.095) (Fig. 3B). Before treatment, the estimated slope difference between the older and the younger age groups was 0.003 (P=0.725), and it was -0.001 (P=0.876) after treatment. The angle of the slope was modified by ERT to a similar degree in both groups (Fig. 3B).
Impact of type of mutation
No significant difference was observed for the height deficit in terms of z-scores between the two groups according the type of mutation (difference in z-score at the start of ERT, -0.705; P=0.666) (Fig. 3C). The angle of the slope was modified by ERT in the patients without severe type of mutation. Before treatment, the estimated slope difference was 0.017 (P=0.158), and it was 0.04 (P<0.001) after treatment (Fig. 3C).
Impact of cognitive involvement
Contrary to expectations, the height deficit was more pronounced in patients without cognitive impairment (difference in z-score at the start of ERT, 1.549; P=0.337) (Fig. 3D). The z-score declined progressively in patients with cognitive involvement; however, a small change was evident in the trajectory of the slope following the initiation of ERT in the patients without cognitive impairment. Before treatment, the estimated slope difference was 0.006 (P=0.623), and it was 0.037 (P<0.001) after treatment (Fig. 3D).
DISCUSSION
This study is the first to report on the effects of ERT on the growth of Asian patients with Hunter syndrome. We evaluated the effects of ERT on the growth of patients from one country who attended a single center. The same method was used to measure the heights of the patients at the center. Therefore, the data are unlikely to be confounded by ethnic differences, and the changes in the heights of the patients were measured relatively accurately. In addition, this study included an analysis of the growth trajectories of Hunter syndrome patients who started to receive ERT before they were 6 yr old.
Patients with MPS II show a significantly different growth pattern to that seen in healthy children. During the first 3 yr of life, MPS II children grow faster than healthy children. However, after this period, they show a clearly negative trend of pathological deviations in comparison with their healthy peers (7). Our patients reflected these two distinct growth patterns found in Hunter syndrome. The patients showed marked growth retardation as they got older, as seen by the height SDS of groups 1, 2, and 3 at the initiation of ERT (0.48±1.71, -2.6±1.79, and -5.12±3.79, respectively).
Today, more patients are diagnosed at a younger age because of the heightened awareness of Hunter syndrome after the approval of ERT for the disease. Six patients were diagnosed at Samsung Medical Center in the last 3 yr. Their median age at diagnosis was 3.1 yr (range 1 yr 2 months-3 yr 8 months), and their height SDS at diagnosis was 0.74±0.99, which reflects the characteristic faster growth of MPS II children during their first 3 yr of life. Among these patients, 3 patients were excluded from this study because their treatment period was less than 2 yr. Evaluating the effect of ERT on the growth of younger aged children with Hunter syndrome is difficult due to their faster growth during their first 3 yr of life compared to the healthy children and their marked growth retardation after this period.
Patients in group 2 showed a tendency toward growth restoration after initiation of ERT, as well as marked growth retardation before treatment initiation. Growth restoration was also seen in the patients in group 3 who started the ERT at over the age of 10 yr. One previous study suggested that ERT may have a positive impact on height, particularly if it was started before 10 yr of age in patients with the attenuated form of the disease (6). In our study, improvements in growth velocity and change of z-score slowed down at 2nd and 3rd year compared to those of at 1st year of ERT in group 2 with attenuated form as shown in Fig. 2A and C. However, their annual growth velocities were improved compared with those before ERT. Interestingly, even the patients who started ERT in their late teens showed an improvement in their growth patterns. This result may be explained by the characteristic delayed puberty of patients with MPS II (although we did not check the pubertal status of the patients in this study), in addition to the large number of individuals (6 of 7 patients) with the attenuated form of the disease.
ERT may have less of an effect on the growth of patients with the severe form because of more severe dysostosis multiplex, malnutrition caused by poor oral intake due to dysphagia, abdominal distension (hepatosplenomegaly), and poor general condition due to multiorgan dysfunction. As shown in Fig. 3D, growth restoration after ERT in the patients with the severe phenotype of the disease was not favorable.
Data from Hunter Outcome Survey (HOS) showed significant improvements in growth after ERT in the study population of 133 patients with both phenotypes of Hunter syndrome aged 8-15 yr at treatment start (15). Similarly, ERT improved the overall growth pattern of the patients in groups 2 and 3 according to their growth curves in this study. In the study from HOS, covariates that significantly impacted on growth were age at start of ERT (8-11 vs 12-15 yr) and type of mutation. In our study, only age at start of ERT (6-10 vs 10-20 yr) had a significant impact on growth.
The improvement in growth observed in this study is likely to have resulted from a reduction in joint contracture rather than new bone growth because ERT is assumed to have little or no effect on bones. Physiotherapy may be beneficial in reducing joint contracture because it can cause the muscles and tendons to adapt to the new bone growth. Most of our patients (26 of the 32) underwent physiotherapy. Other general improvements, such as better pulmonary function, may also have a beneficial impact on the patient's growth.
The z-score, which was used for analysis in this study, is based on the general population, and small numbers of patients with Hunter syndrome could be included in the current study due to rarity of this disease. Information on the natural growth patterns of patients with Hunter syndrome is needed for accurate assessment of the effect of ERT. However, obtaining this type of information is not easy because the disease is very rare, and ethical reasons should dictate that treatment should be offered to all patients diagnosed with Hunter syndrome.
In conclusion, growth in response to ERT could be an important treatment outcome or an endpoint for future studies. Further long-term studies of the effect of ERT on the growth of Asian patients with Hunter syndrome should include larger groups for a more precise evaluation of the effect. In addition, further evaluation is needed of the psychosocial impact of short stature and its amelioration in patients with Hunter syndrome.




 XML Download
XML Download