

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
An increase in the diameter of main pulmonary artery (PAD) on helical computed tomography (CT) is a strong indicator of the presence of pulmonary hypertension. Two recent largest studies by Tan et al. (1) and Edwards et al. (2) suggested that PAD above 2.9 cm and 3.3 cm predicted the presence of pulmonary hypertension. So, CT is used for an indirect method of measurement of mean pulmonary arterial pressure (mPAP) and for a screening method performed by clinician for the presence of pulmonary hypertension (1, 3, 4, 5, 6).
We have anecdotally noted that patients with anthracofibrosis who have a dilated main pulmonary artery on CT scan were often also diagnosed with pulmonary hypertension. Some recent studies have indicated an association between pulmonary hypertension and chronic wood smoke inhalation (7), which has also been shown to cause anthracofibrosis (7, 8). Considering the relationship among chronic wood smoke inhalation, anthracofibrosis, and pulmonary hypertension, we can assume that PAD is increased in the patients with anthracofibrosis. Therefore, we measured PAD of patients with anthracofibrosis and sought to determine the relationship between anthracofibrosis and pulmonary hypertension.
MATERIALS AND METHODS
Patients with anthracofibrosis and CT scans
Between May 2008 and October 2011, a total 71 patients with anthracofibrosis underwent CT scans at our institution at the time of diagnosis. All patients were diagnosed on the basis of two major bronchoscopic criteria, anthracotic pigmentation and bronchostenosis. Of the 71 patients initially identified as having anthracofibrosis, 20 were excluded from this study (8 smokers, 8 patients with tuberculosis, 4 patients with lung cancer). The remaining 51 patients had no history of occupational exposure to mining or industry, congenital or acquired cardiac disease, chronic lung disease, or liver disease. All patients had body mass index <40 kg/m2.
CT scans were obtained using a LightSpeed VCT (General Electric Medical Systems, Milwaukee, WI, USA) with intravenous administration of contrast medium (100 mL at 2-2.5 mL/sec). Scanning parameters included a 130 mA tube current, 120 kV tube voltage, 128×0.6 mm collimation, and 1.2 pitch. All images were reconstructed into axial images with a 5 mm slice thickness at 5 mm intervals.
CT Measurements
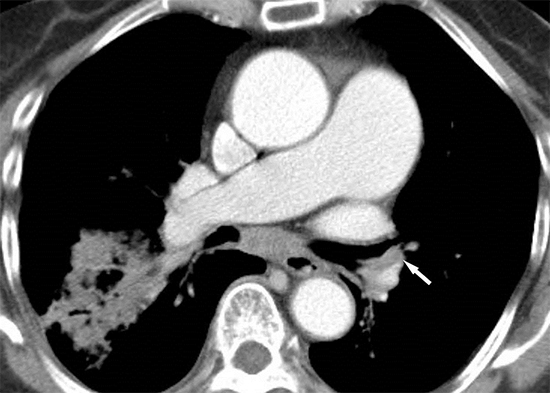
CT images were interpreted independently by two chest radiologists with 2 and 8 yr of experience. The images were viewed in mediastinal windows (window width 350 HU, window level 40 HU). Using an electronic cursor, we measured the widest diameter perpendicular to the long axis of the main pulmonary artery (PAD) at the level of the pulmonary artery bifurcation on a full-screen axial image. We then measured the widest diameter of the ascending aorta (AD) in the same axial image. The outer limits of the contrast medium were used to determine the vessel diameter. Each radiologist measured the PAD and AD twice. The PAD was then divided by the AD to obtain the aortopulmonary ratio (APR). The means of the 4 measurements of the PAD, AD, and APR were used for analysis.
The variation between the intra- and inter-observer measurements was determined using reliability analysis. Mean values were compared with the reference values (one-sample t-test), with 29 mm as the upper reference limit of PAD, as reported by Tan et al. (1). We considered patients with a PAD >33 mm or APR >1 to be at high risk for pulmonary hypertension, and determined the prevalence of anthracofibrosis in this population (2).
Echocardiography
Twenty-one patients underwent echocardiography within 2 months of the initial CT scan. In these patients, 2-dimensional, M-mode pulsed, and color flow Doppler echocardiography was performed by a single examiner. Mean pulmonary arterial pressure (mPAP) was calculated as follows: mPAP=79 - (0.45×Act), where Act (acceleration time) of the pulmonary flow trace is the time interval between the beginning of flow and peak velocity. A mPAP >25 mmHg was used as the cut-off for pulmonary hypertension. We compared the echocardiography criterion (mPAP >25 mmHg) versus CT criteria (PAD >33 mm, APR >1) (McNemar's test) to determine if there was a significant difference between the two tests in diagnosing pulmonary hypertension.
RESULTS
The majority of patients were female (M:F=5:46), and the mean age was 76 yr. The most common symptoms reported were chronic cough, sputum production, and dyspnea.
The reliability test showed no significant differences between intra- and inter-observer measurements within 99% (P<0.001). The mean PAD was 33±5 mm, which was significantly higher than the upper reference limit of 29 mm (P<0.001, one-sample t-test). A total of 30 patients (65%) had a PAD>33 mm, which was considered diagnostic of pulmonary hypertension. The mean AD was 38±4 mm, and the mean APR (PAD/AD) was 0.87±0.12. Only 9 patients (18%) exhibited an APR >1 (Fig. 1, 2 ,3).
Among the 21 patients who underwent echocardiography, 11 (52%) were identified as having pulmonary hypertension. There was no difference in the diagnosis of pulmonary hypertension on CT versus echocardiography using the criteria of PAD >33 mm and mPAP >25 mmHg, respectively (P=1.000, McNemar's test). However, the second CT criterion of APR >1 did not correlate with results obtained on echocardiography (P=0.03, McNemar's test).
DISCUSSION
Anthracofibrosis is characterized by bronchoscopic findings of dark anthracotic pigmentation in conjunction with bronchial narrowing without a relevant history of pneumoconiosis or smoking (9, 10). Anthracotic pigmentations are considered to be mostly the result for carbon particle deposition but iron, lead, silica, phenol, hydrocarbon complexes, and other inorganic or organic substances also can cause this pigmentation (11). Actually, there have been some reports that mixed mineral dusts are associated with anthracofibrosis (12, 13). However, most patients with anthracofibrosis had no history of environmental exposure to coal dust or silica. The epidemiologic, clinical, and bronchoscopic findings of anthracofibrosis are different from those of coal worker's pneumoconiosis and silicosis, which are characterized by particle deposition in small airways and parenchyma with pulmonary fibrosis (11, 12). Patients identified as having this condition are typically older females who have had long-term exposure to wood smoke (11). The mostly widely accepted hypothesis regarding the causes of anthracofibrosis is chronic wood smoke inhalation and tuberculosis. Several recent studies have reported the clinical features as well as radiological and pathological findings of chronic wood smoke inhalation associated with lung disease (CWSLD) (7, 8, 11). Sandoval and co-workers (7) studied 30 patients with pulmonary hypertension and a history of chronic wood smoke inhalation and found that pulmonary arterial pressure was higher in patients with CWSLD than in those with chronic obstructive pulmonary disease (COPD). Additionally, Moran-Mendoza et al. (8) reported that pulmonary hypertension was more common in patients with CWSLD (50%) than in smokers (9%). They concluded that pulmonary hypertension commonly occurs in patients with CWSLD, and that chronic alveolar hypoxia may play an important role in the pathogenesis of pulmonary hypertension in these patients, as it does in patients with COPD. However, they could not identify a reason for the higher incidence of pulmonary hypertension in the CWSLD group compared with the COPD group (7, 8).
Right heart catheterization is the gold standard for diagnosis of pulmonary hypertension (3, 4, 5). This procedure carries significant risks, however, and thus non-invasive approaches are often used for stratification of patients for referral for right heart catheterization. Many studies have shown that CT measurements of PAD strongly correlate with results of right heart catheterization in patients with pulmonary hypertension. Therefore, as a non-invasive test, CT is routinely performed to assess for pulmonary hypertension (1, 2, 6, 8, 14, 15, 16, 17). Most reports have identified, a mean PAD that ranges from 24 to 28 mm, with an upper limit of 29 mm. Thus, a PAD >29 mm is commonly considered to be the CT criterion that determines the presence of pulmonary hypertension (5, 12, 13, 14). However, Edwards et al. (2) and Karazincir et al. (14) reported that a PAD >33 mm was indicative of pulmonary hypertension. They explained that differences in measurement techniques (CT window level, outer limit of main pulmonary artery) and race (Asian versus Anglo-Saxon) could contribute to this discrepancy. In more recent studies, it has been suggested that APR is a more accurate marker of pulmonary hypertension since confounding variables such as patient size influence PAD and AD equally. If the PAD is larger than the AD (APR >1), pulmonary hypertension is usually present (6, 16, 18).
In our study, the mean PAD of patients with anthracofibrosis was statistically larger than the upper limit of normal. This indicates that patients with anthracofibrosis have a significantly dilated main pulmonary artery and an increased pulmonary arterial pressure. Sixty-five percent of our patients even met the CT criterion of a PAD >33 mm as suggested by Edwards et al. (2) and Karazincir et al. (14). Moreover, this result is well-correlated with those obtained using echocardiography, on which 52% of patients were identified as having pulmonary hypertension. This percentage was similar to that reported by Moran-Mendoza et al. (8). However, the mean APR for our patients was less than 1, which did not correlate with the echocardiographic results. This was unexpected since it is discordant with the measurements of the mean PAD. This discrepancy may have been due to age of the patients. Anthracofibrosis occurs primarily in older females, and the mean age of our patients was 76 yr. The diameter of the aorta is known to have significant linear association with age (19, 20). However, the relationship between age and PAD is controversial (2, 14, 17, 18). A dilated AD in older patients may result in a decrease in the APR.
Dilatation of the main pulmonary artery in patients with anthracofibrosis is likely due to chronic alveolar hypoxia, as seen in patients with COPD (7, 8). Considering that bronchostenosis with peribronchovascular fibrotic cuffing is a characteristic finding of anthracofibrosis that differentiates it from COPD, we hypothesize that narrowing of the airways and nearby pulmonary vasculature by the peribronchovascular fibrotic cuff may play an additional role in increasing pulmonary vascular resistance (Fig. 2A, 3B) (9). This finding may also help explain why the tendency of increased pulmonary arterial pressure is higher in patients with anthracofibrosis and CWSLD than in patients with COPD (7, 8, 11).
The major limitation to our study is that none of the patients underwent right heart catheterization. Echocardiographic results can be suggestive of pulmonary hypertension, but this test is not diagnostic. Therefore, it is not certain whether our patients with a PAD >33 mm do in fact have pulmonary hypertension. Therefore, we can not insist that the prevalence of pulmonary hypertension is higher in anthracofibrosis, but that the pulmonary arterial pressure might be increased. In addition, we can not rule out any possibility of being unexpected factors causing pulmonary hypertension or dilatation of the main pulmonary artery in our patient group, although we tried to exclude the factors. Our study may also have been affected by selection bias. Most of our subjects were older females, thus age and gender could have influenced the mean PAD. However, the relationship between these factors and PAD remain controversial. Some studies have reported a tendency toward a smaller PAD in females than in males (2, 14, 17, 18). As a result, the effects of age and gender on our results could be negligible.
In conclusion, patients with anthracofibrosis commonly have a significantly dilated main pulmonary artery and an increased risk of pulmonary hypertension. Although a further investigation about a relationship between pulmonary hypertension and anthracofibrosis is necessary, anthracofibrosis could be one of the causes of pulmonary hypertension, especially in older females, provided that other causative factors have been ruled out.

 XML Download
XML Download