

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Apoptosis is an orchestrated cell death process that is essential for development, maintenance of tissue homeostasis, and the elimination of damaged cells. However, too much or too little apoptosis may lead to diseases such as neurodegenerative disorder, ageing, or cancer. Cancer cells must avoid apoptosis in order for tumors to arise, and defect in the cell death signaling has been regarded as one of the "hallmark" features of cancer. Apoptosis is triggered by the signals that originate from within the cell in the intrinsic apoptotic program or by the signals that originate from outside the cell and activate the death receptors in the extrinsic apoptotic program (1). In the intrinsic death pathway, the Bcl-2 family is comprised of both proapoptotic (Bax, Bak, and Bid) and anti-apoptotic members (Bcl-2, Bcl-xL, and Mcl-1), the imbalance of which influences the susceptibility to apoptosis. Accordingly, it is not surprising that intrinsic apoptotic cascade can become dysregulated during carcinogenesis. Indeed, dysregulation of the Bcl-2 family is common in many human cancers and may contribute to the development of cellular resistance to chemotherapeutic drugs, as upregulation of these proteins confers a survival advantage that prevents cell death in response to a variety of apoptotic stimuli (2, 3).
The importance of the anti-apoptotic proteins Bcl-xL and Mcl-1 on drug resistance and relapse has been increasingly recognized (4, 5). Downregulation of Bcl-2, Bcl-xL, or Mcl-1 by siRNA-mediated gene silencing sensitizes cells to chemotherapy (6), whereas their high expression has been associated with increased drug resistance and poor prognosis in several types of cancers (5, 7). Bcl-xL and Mcl-1, but not Bcl-2, have also been shown to cooperatively protect cells from death stimuli by direct sequestration of proapoptotic protein Bak (3, 8). These observations indicate that simultaneous loss of Bcl-xL and Mcl-1 rather than that of Mcl-1 alone is needed to induce Bak-mediated apoptosis. New inhibitors, including gossypol and obatoclax, have been developed to target the anti-apoptotic roles of Bcl-xL and Mcl-1 in a variety of human malignancies (9, 10). In malignant mesothelioma (MM), Bcl-xL and Mcl-1 have been shown to be highly expressed as compared to Bcl-2 (11), suggesting that its anti-apoptotic phenotype could be associated with these two key anti-apoptotic factors rather than Bcl-2. Therefore, anti-apoptotic strategies, such as dual inhibition of Bcl-xL and Mcl-1, are expected to play a potentially important role in cancer therapy, in particular against chemoresistant cancers.
MM is an asbestos-related tumor that arises from the mesothelial cells lining the pleural, pericardial, and peritoneal cavities. The prognosis is very poor with a median survival time of approximately 9.8 months from the time of diagnosis (12). Despite recent advances in the diagnosis and therapy, intrinsic or acquired resistance and poor response rates to currently available anticancer drugs remain responsible for failure of chemotherapy in MM. The highly resistant phenotype of this tumor to apoptosis is a main reason for chemoresistance and contributes to therapy failure and tumor recurrence (13). Therefore, targeting anti-apoptotic proteins may provide a promising strategy to overcome the resistance to chemotherapy-induced cell death.
Recently, we previously reported an in vitro synergistic model of resveratrol and chemotherapeutic agent clofarabine using a MM cell line, MSTO-211H. The molecular mechanisms that mediate such synergistic actions of two compounds have been characterized and included simultaneous targeting of multiple biological pathways involving activation of p53 (14), reduction of Nrf2 activity (15), and suppression of Sp1 and PI3-kinase/Akt survival proteins (16). Here, using MM H-2452 cell line showing a mild sensitivity against co-treatment with resveratrol (Res) and clofarabine (Clo), we investigated the significant roles of anti-apoptotic molecules involved in this process with a final goal for better understanding the molecular mechanism of the relative resistance to Res plus Clo. This study reports that Res plus Clo, which reduce Mcl-1 protein level, elicited the greater potency for killing and inhibiting the growth of H-2452 cells exposed to Bcl-xL-specific siRNA.
MATERIALS AND METHODS
Reagents and cell culture
Resveratrol, clofarabine, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrozolum bromide (MTT), sodium dodecyl sulfate (SDS), phosphate buffered saline (PBS), and antibody to β-actin were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA). Trizol reagent, Mcl-1- and Bcl-xL-targeting small interfering RNAs (siRNAs), cell culture media and reagents were purchased from Invitrogen (Carlsbad, CA, USA). Anti-human Mcl-1, Bcl-xL, caspase-3, cleaved caspase-3, and poly (ADP-ribose) polymerase (PARP) antibodies were from Cell Signaling Technologies (Beverly, MA, USA). Antibodies to horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The human mesothelioma cell line H-2452 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). H-2452 cells were maintained in RPMI-1640 medium supplemented with 5% fetal bovine serum, 1 mM glutamine, 100 units of penicillin/mL and 100 µg of streptomycin/mL. Cells were grown to 50% confluence in a monolayer culture in this medium for 24 hr before treatment.
Cell proliferation assay
The cells were seeded in 96-well microtiter plates, and were treated with drugs or chemicals at different concentrations for the indicated times, after which they were exposed to MTT (final 0.1 mg/mL) for additional 4 hr. The formazan crystals, formed by the reduction of MTT in living cells, were solubilized in 200 µL of DMSO and were measured with spectrophotometry at 560 nm. The results were expressed as percentage, based on the ratio of the absorbance between the treated cells and the controls (100%).
Western blot analysis
Cell lysate containing 40 µg of protein was separated on NuPAGE 4%-12% bis-tris polyacrylamide gels (Invitrogen, Carlsbad, CA, USA) and were electrophoretically transferred to Immuno-Blot PVDF membranes. The signal was visualized by an enhanced chemiluminescence kit (Santa crutz), using X-ray films. The blots were then stripped with a stripping buffer (100 mM β-mercaptoethanol, 2% SDS, and 62.5 mM Tris-HCl, pH 6.7) and were reprobed with anti-β-actin antibody as a loading control.
Reverse transcription-polymerase chain reaction (RT-PCR)
One µg of the total RNA was converted to cDNA using the oligo (dT)15 primer and AMV reverse transcriptase (iNtRON Biotechnology, Seongnam, Korea). Each single-stranded cDNA was then diluted and subjected to PCR amplification using i-MAX II™ DNA polymerase (iNtRON Biotechnology). The PCR conditions were as follows; initial denaturation at 94℃ for 2 min was followed by 28 cycles of 94℃ for 40 sec, 57℃ for 40 sec and 72℃ for 40 sec. The following primers were used to amplify the fragments of the genes. Specific primers for RT-PCR amplification of various genes were as follows: (a) Mcl-1: sense 5'-cggtaatcggactcaacctc-3' and antisense 5'-cctccttctccgtagccaa-3', (b) Bcl-xL: sense 5'-ggctgggatacttttgtaga-3' and antisense 5'-aagagtgagcccagcagaac-3', (c) GAPDH: sense 5'-acctgacctgccgtctagaa-3' and antisense 5'-tccaccaccctgttgctgta-3'. The PCR products were resolved by electrophoresis in a 1.5% agarose gel containing ethidium bromide and bands were visualized under UV light.
RNA interference
RNA interference was performed using siRNAs targeting Mcl-1 (Oligo ID: HSS181041) and Bcl-xL (Invitrogen, Oligo ID: HSS141361). Briefly, the cells were seeded in 96-well or 6-well plates and were transfected at 40% confluency with siRNA duplex using a lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's recommendations. Cells transfected with the Stealth RNAi negative control duplex (Invitrogen) were used as controls. After 24 hr of transfection, cells were treated with DMSO or with Res plus Clo.
Caspase-3/7 activity
Activation of caspase-3/7 was quantified with the ApoTox-Glo™ Triplex Assay kit according to the manufacturer's protocol (Promega). Cells were seeded in 96-well microtiter plates and were treated with Res plus Clo for the indicated times. Cells were incubated with substrate containing Caspase-Glo® 3/7 assay buffer for 30 min. Caspase-3/7 activities were calculated after detection of luminescence by a GloMax-Multi Microplate Multimode Reader. The results were expressed as percentages, based on the ratios of the luminescence of the treated cells to that of the controls (100%).
Apoptosis assay
The apoptotic cell distribution was determined using a Muse™ Annexin V & Dead Cell kit according manufacturer's protocol (Merck Milipore, Darmstadt, Germany). Briefly, cells were collected in culture medium, mixed with the Muse Annexin V and Dead Cell Reagent, and analyzed using a Muse Cell Analyzer (Merck Milipore).
Cell cycle analysis
Trypsinized cells were pelleted by centrifugation and fixed in 70% ice-cold ethanol overnight at -20℃, and treated with DNase-free RNase A (150 µg/mL) and propidium iodide (20 µg/mL). DNA content was analyzed by a MACSQuant Analyzer (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) according to fluorescence of propidium iodide (PI).
Statistical analysis
Statistical comparisons were performed using one-way analysis of variance (ANOVA) followed by a Tukey's post hoc correlation for multiple comparisons using SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA). Data were expressed as the mean±SEM. Significant differences were considered with values of P<0.05.
RESULTS
Resveratrol and clofarabine downregulate Mcl-1 protein level with modest inhibition of H-2452 cells proliferation
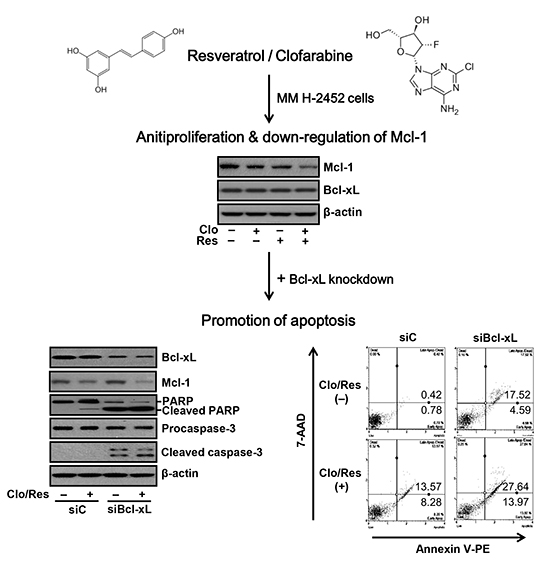
To determine subtoxic doses for the treatment of Res plus Clo, the effects of continuous exposure to various concentrations of two compounds on H-2452 cells were initially assessed by MTT assays. During the 72 hr exposure to Clo (0-320 nM) or Res (0-30 µM), cell proliferation was significantly inhibited in a dose-dependent manner (Fig. 1A). However, Res up to 15 µM was non-toxic (IC10=17 µM), while the next higher dose of 20 µM significantly inhibited cell growth. Next, H-2452 cells were co-treated for 72 hr with increasing concentrations of Clo (20, 40, 80, 160, and 320 nM) and Res (15 µM) and the inhibition of cell proliferation was assessed using the MTT assay. Clo treatment at the indicated concentrations reduced cell proliferation by approximately 9.9%, 12.8%, 17.2%, 24.8%, and 40.5%, respectively (Fig. 1B). However, the combination with Res inhibited cell proliferation by approximately 19.6%, 29.6%, 32.5%, 39.1%, and 48.9%, respectively. Additionally, we studied the kinetic effect of continuous exposure to Res and Clo, alone or in combination, on H-2452 cells at the same concentration as we used in previous experiments in which a marked synergistic cytotoxicity of another human MM cell line, MSTO-211H, was observed (14). Cells exposed to Res (15 µM) and Clo (40 nM) showed a mild antiproliferative effect from 24 hr to 72 hr. As shown in Fig. 1C, 72 hr after treatment, the percent inhibition of cell proliferation was approximately 29.6% of that of control cells in H-2452 cells, whereas approximately 55% was inhibited in MSTO-211H cells in our previous experiments. To understand the molecular basis of the growth-inhibitory mechanism caused by Res plus Clo, we evaluated the effect of the combination treatment on anti-apoptotic proteins, Mcl-1 and Bcl-xL. Western blot analysis showed that Mcl-1 protein levels were downregulated following treatment (Fig. 2A) and were maintained at low levels over 72 hr (Fig. 2B). In combination with Clo, the ability of Res to reduce the contents of Mcl-1 was also confirmed in a dose-dependent experiment (Fig. 2C). However, significant changes in the Bcl-xL protein level were not found in this condition.
Mcl-1 protein level, in response to resveratrol and clofarabine, is regulated by proteasomal activity
To elucidate the possible mechanism underlying Mcl-1 downregulation by Res plus Clo, cells were co-treated with two compounds for 24 hr. As shown in Fig. 3A, there were no detectable changes in the Mcl-1 mRNA levels in RT-PCR analysis. These data suggest that Mcl-1 protein levels in H-2452 cells in response to Res plus Clo were regulated through transcription-independent mechanism. In contrast, the amount of Mcl-1 protein upon the combination treatment significantly increased following the pretreatment with proteasome inhibitor MG132 (Fig. 3B). Next, we analyzed the effect of protein synthesis inhibitor, cycloheximide (CHX), on Res plus Clo-induced downregulation of Mcl-1. The Mcl-1 protein level was gradually declined over 160 min of treatment with CHX alone, suggesting rapid turnover of Mcl-1. However, in the presence of MG132 and CHX, the Mcl-1 protein level was retained to a greater extent compared to cells treated with CHX alone, suggestingg that Mcl-1 was stabilized by proteasomal activity (Fig. 3C). These results indicate that Mcl-1 protein level, in response to Res plus Clo, was mainly regulated at the posttranslational step in H-2452 cells.
Co-silencing of Mcl-1and Bcl-xL induces G2/M cell cycle arrest and caspase-dependent apoptosis
To examine whether dual inhibition of Mcl-1 and Bcl-xL sensitizes H-2452 cells to Res plus Clo, cells were transfected with Bcl-xL siRNA (siBcl-xL) or in combination with Mcl-1 siRNA (siMcl-1) before exposure to two compounds. As shown in Fig. 4A, knockdown of Bcl-xL gradually inhibited cell proliferation over 72 hr as determined by MTT assay. These responses were augmented following a subsequent exposure to Res plus Clo. In flow cytometric analysis, the sub-G0 peak, indicative of apoptosis, significantly increased when cells were knocked down with siBcl-xL. A strong sub-G0 peak was observed in cells co-transfected with siMcl-1 plus siBcl-xL (Fig. 4B). In the order of siMcl-1 plus siBcl-xL>siBcl-xL>siC, the percentage of cells in the G0/G1 and S phases tended to decrease and that in the G2/M phase tended to increase, indicative of an cell cycle arrest (Fig. 4C). Consistently, knockdown of Bcl-xL induced an increase in the caspase-3/7 activity (Fig. 5A) with enhanced cleavages of procaspase-3 and its substrate PARP (Fig. 5B), compared to those of the control siRNA (siC). The occurrence of the G2/M arrest was accompanied by an increase in the percentage of apoptotic cells, determined by annexin V binding assay (Fig. 5C). These findings became apparent following exposure of cells to Res plus Clo.
DISCUSSION
Anti-apoptotic proteins of Bcl-2 family members, such as Bcl-2, Mcl-1, and Bcl-xL, are frequently dysregulated during carcinogenesis and their increased expression has been reported to protect cells from apoptotic stimuli in numerous human cancer types (2, 7). Some therapeutic approaches have also been reported to be aimed at suppressing expression or activities of these anti-apoptotic proteins as an effective strategy for overcoming the drug resistance in cancers. Compared to MSTO-211H cells, in which two compounds had caused massive cell death in our previous study (14), Res plus Clo were less efficient in killing H-2452 cells. Investigating the underlying mechanism(s) of H-2452 cells showing a mild sensitivity to the combination treatment, we found that Res plus Clo downregulated Mcl-1 protein level without any significant change on Bcl-xL protein level. From this point of view, we supposed that the compensatory action of anti-apoptotic Bcl-xL protein, possibly due to its high endogenous level, may be required for increasing a limited cytotoxicity in H-2452 cells. This observation prompted us to further determine whether cellular levels of Mcl-1 and Bcl-xL would have influence on Res plus Clo-induced lethality.
The Mcl-1 protein level has been regulated through several different mechanisms at the transcriptional, translational, and posttranslational steps depending on the cell types and culture conditions (17). The ability of cells to regulate Mcl-1 protein level at multiple steps reflects its importance as the means for preventing apoptosis and cell death against endogenous and/or exogenous cytotoxic insults. In the present study, Res plus Clo reduced Mcl-1 protein level without any change in its mRNA level. Instead, treatment of H-2452 cells with proteasome inhibitor promoted Mcl-1 accumulation to the levels far above the basal levels and prolonged its half-life by inhibition of proteasomal degradation. This finding suggests that change in Mcl-1 stability is one of the possible mechanisms explaining this discrepancy.When H-2452 cells treated with MG132 followed CHX to block the de novo synthesis of Mcl-1 protein, its half-life became longer than 2 hr. In contrast, cells treated with CHX alone were shown to have a half-life of about 16.5 min. The build-up of Mcl-1 protein in cells upon inhibition of MG-132 is in agreement with earlier observations from human neutrophil (18) and human Jurkat cells (19).
During apoptosis, Mcl-1 induction tends to be downregulated and its depletion contributes to activating the intrinsic apoptotic pathway (20). In previous studies, suppression of Mcl-1 expression by RNA interference has been described to promote apoptosis induced by chemotherapeutic agents in several cancer types (21, 22). However, recent evidence proposed that Mcl-1 and Bcl-xL cooperatively act to prevent activation of proapoptotic protein Bak. They have shown that inhibition of a possible cooperation with Mcl-1 and Bcl-xL is required for Bak-induced apoptosis (23). Therefore, it is reasonable to suppose that high endogenous level of Bcl-xL may reduce the effects of Res plus Clo by attenuating activities of proapoptotic proteins Bax and Bak. This interpretation is supported by results showing that the co-silencing of Mcl-1 and Bcl-xL chemosensitizes cells to various apoptotic stimuli in several cancer models (4, 24). Consistent with these findings, our data that simultaneous downregulation of Mcl-1 (by Res plus Clo) and Bcl-xL (by RNA interference) displayed an enhanced sensitivity to apoptosis further highlights the importance of targeting Bcl-xL in this process.
The notion of cooperativity between Mcl-1 and Bcl-xL was also observed in the cell cycle distribution of H-2452 cells. Dual inhibition of Bcl-xL and Mcl-1 induced cell accumulation in the G2/M phase, which is critical for growth inhibition, with a concomitant detection of the sub-G0/G1 peak. A similar activation of G2/M arrest and apoptosis has also been reported in fisetin-treated human epidermoid carcinoma cells, which showed an increased sensitivity to apoptosis with reduced expression of Bcl-xL and Mcl-1 (25). DNA damage results in cell cycle arrest at the G1 or G2 phase through mechanisms involving the G1 or G2 phase checkpoint, respectively, in which damaged cells stop DNA replication and mitosis, presumably allowing cells to repair DNA lesions or to undergo apoptosis before cell cycle pregression to next round. Although our study did not provide direct evidence that apoptosis is occurring in the cells arrested in G2/M phase, the data indicate that Bcl-xL, in cooperation with Mcl-1, may be necessary for phase-specific cell cycle regulation in H-2452 cells. The role of these molecules in the regulation of the cell cycle has been undertaken in our laboratory.
In conclusion, our data have shown that the simultaneous targeting of Mcl-1 and Bcl-xL induced strong anti-cancer effects for inhibiting cell proliferation, for promoting apoptosis, and for blocking cell cycle progression in MM H-2452 cells showing a mild sensitivity to Res plus Clo. These results demonstrate a potential significance of dual targeting of Mcl-1 and Bcl-xL as a promising strategy for overcoming resistance to these compounds. Pharmacologic approaches that counteract Bcl-xL accumulation may increase the efficacy of these compounds. Further studies on animal models are required to determine if this strategy may have potential in humans as an effective anticancer agent for MM.





 XML Download
XML Download