

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Neuropathic pain is a common consequence of damage to the nervous system via the mechanisms of infection, nerve injury, diabetes, and so forth (1). Many recent studies have employed diverse animal models of neuropathic pain, and indicate that the abnormal excitability of dorsal horn neurons and the activation of spinal microglia by peripheral sensory input influence the induction of neuropathic pain (2, 3, 4, 5).
Charcot-Marie-Tooth disease (CMT) is a hereditary motor and sensory neuropathy and one of the most common hereditary neuromuscular diseases with a worldwide prevalence of 1/2,500 people (6). CMT is a genetically heterogeneous group of disorders and is induced by mutation of genes, which leads to length-dependent axonal degeneration. There are two main subdivisions of CMT: CMT1 (demyelinating form) and CMT2 (neuronal form). This disease is characterized by the loss of somatosensation across various parts of the body and progressive muscle atrophy in the extremities. According to CMT patients, neuropathic pain is often a symptom of CMT (7, 8, 9), along with severe pain that interferes with quality of life.
Glycyl-tRNA synthetase (GARS) catalyzes the ligation of glycine to its cognate tRNA and is ubiquitously expressed in both neuronal and non-neuronal cells. GARS gene mutations have been found to influence GARS axonopathies (10). To date, several mutations of GARS have been reported and, of these, L129P and G240R are the most closely linked to GARS axonopathies such as CMT type 2D (CMT2D) and distal spinal muscular atrophy type V (dSMA-V) (10, 11, 12, 13). However, there are no pharmacological or genetic treatments for this disease.
Here, for the first time, a mouse model of neuropathic pain (CMT2D and dSMA-V) using GARS wild type (WT) and L129P, and G240R mutant-expressing adenovirus vectors is described. Several pain-related markers, including p-ERK, activating transcription factor 3 (ATF3) and Iba1 (14, 15, 16, 17), were evaluated in the dorsal horn of the spinal cord and dorsal root ganglion (DRG) neurons to confirm the anatomical phenotypes of neuropathic pain in this GARS-induced mouse model of axonopathy. This model of CMT2D and dSMA-V using an adenovirus vector may help to identify the underlying mechanisms of neuropathic pain in CMT.
MATERIALS AND METHODS
Animals
All experiments were performed using 6-week-old male C57BL/6 mice, which were obtained from Semtako (Osan, Korea). All subjects were housed in a temperature- and humidity-controlled environment on a 12-hr light/dark cycle, provided food and water ad libitum, and had their cages were changed weekly.
Neuron-specific recombinant adenoviruses
The mouse choline acetyltransferase (ChAT) gene (GenBank accession number NC_000080) was used as the promoter, and included the neuron-restrictive silencer element (NRSE) and a cholinergic-specific enhancer, which are key regulatory elements for neuron-specific expression (18, 19). The use of human GARS (hGARS) clones allowed for subcloning into the pAdTrack-ChAT-CMV/GFP vector. The E1/E3 double-deleted supercoiled adenoviral backbone vector (pAdEasy-1; Stratagene; Santa Clara, CA, USA) was used to generate stable homologous recombinants with pAdTrack-ChAT-hGARSwt-CMV/GFP, pAdTrack-ChAT-hGARSL129P-CMV/GFP, and pAdTrack-ChAT-hGARSL129P-CMV/GFP (Fig. 1A). The titers of Ad/ChAT, AdhGARSwt/ChAT, AdhGARSG230R/ChAT, and AdhGARSL129P/ChAT employed in these experiments were 3.2×109, 5.1×109, 4.9×109, and 2.6×109 pfu/mL, respectively.
Viral administration procedures
Recombinant adenoviruses were injected into the sciatic nerve. Mice were anesthetized with 30 mg/kg Zoletil (Virbac; Carros Sedecs, France) and 10 mg/kg Rompun (Bayer; Leverkuzen, Germany) before the sciatic nerve was surgically exposed via an incision of the musculus gluteus superficialis. This virus diluent of AdhGARSwt/ChAT (1.5 µL), AdhGARSG204R/ChAT (1.5 µL), or AdhGARSL129P/ChAT (3 µL) containing 0.1% fast green as a tracer was then injected into the sciatic nerves using a Hamilton syringe with a glass micropipette under a stereoscope. The skin incision was closed with Silkam 4.0 sutures (B/Braun; Melsungen, Germany).
Immunofluorescence staining
DRG neuron and spinal cord tissue slides were fixed in 4% paraformaldehyde (PFA) for 10 min. Following three washes in phosphate-buffered saline (PBS), the slides were permeabilized in PBS containing 0.3% Triton X-100 (PBST) and blocked with 10% bovine serum albumin (BSA) overnight at 4℃. The slides were then incubated with the primary antibodies; FLAG (1:500; Cell Signaling Technology; Cat #: 2368, Danvers, MA, USA), Iba1 (1:1,000; Wako Pure Chemical Industries Ltd.; Cat #: 019-19741, Osaka, Japan), and ATF3 (Santa Cruz Biotechnology; Cat #: sc-188, Dallas, TX, USA) for 16 hr at 4℃. Following three washes in PBS, the slides were incubated for 2 hr at room temperature in the Alexa Fluor 594 secondary antibody (1:1,000; Invitrogen, Life Technologies; Madison, WI, USA). The slides were then washed with PBS and mounted. Immunofluorescence was detected and viewed using a laser scanning confocal microscope (LSM700; Carl Zeiss; Oberkochen, Germany).
Western blotting
The DRG neurons or spinal dorsal horn tissues were lysed in a modified radioimmune precipitation assay buffer (RIPA; 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA, 0.5% deoxycholic acid, 2 µg/mL aprotinin, 1 mM phenylmethylsulfonyl fluoride, 5 mM benzamidine, 1 mM sodium orthovanadate, and 1×protease inhibitor cocktail [Roche; Indianapolis, IN, USA]). Protein extracts were separated using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then electrotransferred onto a nitrocellulose membrane. After blocking with 5% non-fat milk in Tris-buffered saline (TBS) containing 0.05% Tween 20 (TBST) overnight, the membrane was incubated with the following primary antibodies for 1 hr at room temperature; mouse anti-beta actin (1:5,000; Sigma; St. Louis, MO, USA), rabbit anti-FLAG (1:1,000; Cell Signaling Technology), and p-ERK1/2 (1:1,000; Cat #: 9101, Cell Signaling Technology). After three 10-min washes in TBST, the blots were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (1:2,000; Invitrogen; Carlsbad, CA, USA). Signals were detected using an enhanced chemiluminescence reaction (Amersham; Buckinghamshire, England). All experiments were repeated at least three times.
Reverse transcription polymerase chain reaction
Total RNA was obtained from DRG neuron and spinal dorsal horn tissues from either side 7 days following adenovirus transfection into sciatic nerves using the acid quanidine iso-thiocyanate/phenol/chloroform extraction (AGPC) method. First-strand complementary DNA (cDNA) was converted from total RNA with SuperScript II (Invitrogen) and used for reverse transcription polymerase chain reaction (RT-PCR) amplification. The thermal cycling conditions were 94℃ for 5 min, 28-33 cycles of amplification consisting of 94℃ for 30 sec, 55℃ for 30 sec and 72℃ for 30 sec before a final cycle at 72℃ for 7 min. The designed primer sets were as follows: GAPDH-Forward- : 5'-CAGCAATGCATCCTGCACC-3', GAPDH-Reverse- : 5'-TGGACTGTGGTCATGAGCCC-3', FLAG-Forward- : 5'-ACGC GTCGACATGGACGGCGCGGGGGCTGA-3', FLAG -Reverse- : 5'-AGCC GGTACC TCA CTTTGTCATCGTCATCCTTGTAATC TTCCTCGATTGTCTCTTTTT-3'. Each reaction was performed in triplicate and the results were visualized using 1% agarose gel electrophoresis stained with GelRed (Biotium; Hayward, CA, USA).
Behavioral testing
Animals were habituated to the experimental room for 2 hr before behavior testing. To assess foot mechanical sensitivity, calibrated von Frey filaments (1.0-4.0 g; Stoelting; Wood Dale, IL, USA) were applied to the plantar surfaces of the hindpaws. The 50% paw withdrawal threshold (PWT) was measured using Dixon's up-and-down method (20).
Statistical analysis
Differences in the means between groups were subjected to an analysis of variance (ANOVA) followed by a Bonferroni post-hoc test to correct for multiple comparisons. P values of <0.001 were set as the levels of statistical significance for all tests.
Ethics statement
All animal procedures were performed in accordance with guidelines of the Korean Academy of Medical Science and approved by the Kyung Hee University Committee on Animal Research (KHUASP[SE]-12-021). Every effort was made to minimize the suffering of the animal and the number of animals used.
RESULTS
Based on previous research, it is known that the GARS mutant proteins L129P and G240R are involved in dSMA-V and CMT2D, repectively (10, 12, 21). To develop CMT2D and dSMA-V animal models of neuropathic pain, an adenovirus vector system were employed. Recombinant adenoviruses (pAdeasy-ChAT-hGARS-FLAG-CMV-GFP) that expressed the FLAG-tagged hGARS protein under the control of the cell-type-specific ChAT promoter and the green fluorescent protein (GFP) gene under the control of the general CMV promoter were used to transfect the sciatic nerves of mice to construct WT and L129P/G240R mutant hGARS-overexpressing mouse models (Fig. 1A). Around the injected site, both Schwann cells and peripheral axons were transfected by adenovirus vectors and expressed the GFP fluorescence, but FLAG was only expressed in peripheral axons due to the neuron-specific promoter (data not shown). However, in the spinal cord, the GFP fluorescence was only expressed in the peripheral projection-related neurons, but not spinal glial cells. To assess the expression of adenovirus vectors in the spinal cord, immunolabeling was performed using an anti-FLAG antibody to detect the expression of transfected WT, L129P and G240R GARS genes. Because GFP in an adenovirus vector is a separate transcript driven by the CMV promoter, all adenovirus vectors (WT, L129P, G240R and an empty vector without GARS gene) exhibited GFP-positive signals in the spinal cord (Fig. 1B). Fig. 1B also indicated the transfection was equally accomplished in the both GARS WT- and mutant-expressing sides similar to loading control using β-actin in western blotting. However, the patterns of anti-FLAG immunostaining in the spinal cord are different for each adenovirus vector. Because the empty virus vector contains only the ChAT promoter, it cannot express GARS-FLAG fusion protein as do the other adenovirus vectors (Fig. 1B); WT-, G240R- and L129P-expressed portions of the spinal cord exhibited anti-FLAG signals in neuronal cell bodies. Additionally, immunostaining for FLAG demonstrated the presence of GFP/FLAG double positive axons, which indicates that the adenovirus vectors effectively expressed GARS proteins (Fig. 1A). Thus, these findings suggest neuron-specific expression of the FLAG-tagged hGARS fusion protein in peripheral neurons.
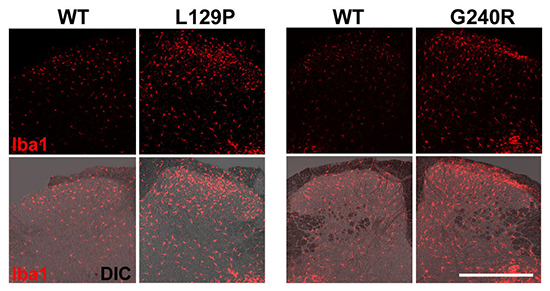
Recent studies have demonstated the role of activated microglia in chronic neuropathic pain (5, 22, 23). When allodynia is apparent, the number of microglia on the injured side of spinal cord identified by microglial markers increases dramatically (4, 17). To evaluate the spatial extent of these activated microglia relative to the mutant GARS protein in the spinal dorsal horn, Iba1 immunoreactivity, as a marker of activated microglia, was compared between WT GARS- and mutant GARS-expressing portion of the spinal dorsal horn. A significant increase in the number of microglia was observed in the L129P and G240R mutant GARS-expressing portions relative to the WT GARS-expressing portion (Fig. 2A, B). To minimize the differences in the expression of each viral vector-induced GARS protein (WT, L129P, and G240R), the differences were compensated by determining GFP expression by means of assaying Iba1 expression (Fig. 2B). Thus, these findings indicate that the expression of mutant L129P and G240R GARS in the central axon terminals of the spinal cord increases the number of activated microglia, which is sufficient to induce neuropathic pain in a model of mutant GARS-induced neuropathy.
p44/42 MAPK (ERK1/2) is a member of the MAPK family that plays a critical role in intracellular signal transduction. Following peripheral nerve damage, microglia in the spinal dorsal horn express ERK, which contributes to the induction of neuropathic pain (24, 25). To determine whether ERK activation is involved in the mutant GARS-expressing portions of the spinal dorsal horn, Western blotting was performed on spinal dorsal horn samples using anti-pERK1/2 (Fig. 3A). A significant activation of ERK1/2 in the mutant GARS-expressing dorsal horn samples was observed compared to WT GARS-expressing samples (Fig. 3A). Both pERK1 (p44 MAPK) and pERK2 (p42 MAPK) levels were increased significantly in the mutant GARS expressing dorsal horn compared with WT GARS-expressing samples (Fig. 3B). To investigate the role of GARS mutation in the pathogenesis of pain hypersensitivity, we compared the pain behaviors of WT-, Empt-, G240R-, and L129P-expressing mice. Immediately after the sciatic nerves were infected with the adenovirus, WT, Empt, G240R, and L129P mutant hGARS-expressing mice showed a significant decrease in PWT (Fig. 3C). Over time, however, the WT-GARS- and Empt-expressing mice exhibited significantly improved PWT compared with the G240R- and L129P-expressing mice and the allodynia behavior in G240R- and L129P-expressing mice was unchanged for 7 days after adenovirus infection (Fig. 3C). In addition, the duration of the pain in this model is less than 3 weeks (Fig. 2C) because of in vivo immune reaction to adenovirus. Thus, these results suggest that mutant GARS (L129P and G240R) expression in axon terminals of the spinal dorsal horn may mediate ERK1/2 activation and be involved in the development of neuropathic pain.
To identify the pathophysiological mechanisms underlying neuropathic pain, several studies have monitored the induction of ATF3, a selective marker of nerve injury (14, 15, 26). These studies found that stimuli in the peripheral nerves induce the expression of ATF3 in DRG neurons in animal models of neuropathic pain, suggesting that ATF3 has a significant influence on neuropathic pain. Here, ATF3 was also utilized as a possible marker of sensory damage that affects neuropathic pain in mutant GARS-induced neuropathies. A previous report found that immunohistochemically stained ChAT could be visualized in the myelinated A-fibers and unmyelinated C-fibers of the adult rat DRG neurons (27, 28). To identify the expression of GARS fusion proteins in DRG neurons, the expression of the viral protein FLAG was examined in DRG tissues by Western blotting and RT-PCR (Fig. 4A, B). FLAG expression was confirmed in DRG neurons following viral vector transfection into the sciatic nerves (Fig. 4A, B). To assess ATF3 activation in mutant-GARS-expressing DRG neurons compared with WT-GARS-expressing neurons, DRG tissues were immunostained with an anti-ATF3 antibody. ATF3 staining identified an increased amount of mutant-GARS-expressing (L129P and G240R) DRG neurons compared with WT-GARS-expressing neurons (Fig. 4C). ATF3 staining was strong in the area detected by GFP green fluorescence in mutant-GARS-expressing DRG neurons, but no ATF3-positive signals were detected in WT-GARS-expressing DRG neurons (Fig. 4C). Quantitative analyses also revealed an increase in the number of ATF3-positive signals in the area positive for GFP fluorescence in mutant-GARS-expressing DRG neurons compared with WT-GARS-expressing DRG neurons. Thus, these data suggest that mutant GARS protein expression in DRG sensory neurons may be involved in ATF3 activation and that the central axon terminals of DRG neurons associated with ATF3 activation may affect the recruitment of microglia and contribute to the initiation of neuropathic pain in mutant GARS-induced neuropathies.
DISCUSSION
Interestingly, previous studies have shown the in vitro and in vivo distribution defect of GARS mutants in the axons, but not cell body (29, 30). In Fig. 2A, middle two high magnification images showed that the expression of GFP fluorescence in the dorsal horn was only accomplished in the GARS WT-expressing central processes of DRG neurons whose peripheral axons were infected by adenovirus vectors, but did not in the GARS mutant-expressing dorsal horn.
Damage to the peripheral nervous system induced by CMT can result in neuropathic pain, which is difficult to manage effectively. To date, studies of CMT have focused primarily on basic genetic analyses related to the cause of this disease and on some clinically relevant symptoms; thus, less is known regarding the important role of neuropathic pain. However, a growing amount of evidence indicates that pain in CMT is occasional and that the clinically pain is most often neuropathic (7, 8, 9). We developed the animal model of CMT-related neuropathic pain (CMT2D and dSMA-V) using adenovirus vectors. Following the transfection of GARS WT-, L129P-, and G240R-expressing virus vectors into the sciatic nerve, the equal expression of GARS WT, L129P, and G240R fusion proteins was confirmed via FLAG-tag expression in each portion of the spinal dorsal horn (Fig. 1B). Additionally, this study demonstrated that GARS mutants are morphologically involved in neuropathic pain using Iba1, ATF3, and pERK1/2, possible markers of neuropathic pain. Although further evaluation is necessary to optimize this animal model of neuropathic CMT pain, it seems likely that this type of model may be able to explain the morphological phenotypes of neuropathic pain associated with CMT. Therefore, this animal model may help to identify the molecular mechanisms underlying neuropathic pain in CMT diseases and represent a new approach to discovery of therapeutic targets.



 XML Download
XML Download