

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Type 1 diabetes (T1D) is one of the classical examples of organ-specific autoimmune diseases characterized by autoimmunity against β-cell autoantigens. Recent papers have reported that the initial event in the pathogenesis of autoimmune T1D comprises sensing of death-associated molecular patterns (DAMPs) released from apoptotic β-cells by innate immune receptors such as Toll-like receptor (TLR), which suggests that innate immunity is critical for the initiation and development of T1D. In contrast to T1D, type 2 diabetes (T2D) has been regarded as a metabolic disorder that is pathogenetically different from T1D for a long time. However, recent investigations have revealed a significant contribution of chronic inflammation in the pathogenesis of T2D. While it is not entirely clear which innate receptors are critical in T2D, recent papers have shown an important role for NLRP3 in metabolic syndrome associated with lipid injury. In this review, the role of innate immunity in the pathogenesis of both T1D and T2D will be covered in an attempt to find common ground for the two diseases.
ROLE OF INNATE IMMUNITY IN T1D
TLR2 as a sensor of β-cell death in T1D
In T1D as a representative organ-specific autoimmune disease, specific immunity is critical considering the disease association with specific HLA types, the presence of specific autoantibodies to specific diabetic autoantigens, and the development of diabetes after transfer of specific autoreactive T cells, etc. However, recent investigations by many investigators showed essential role of innate immunity also which has not been appreciated for a long time. Indeed, the field of innate immunity itself has been neglected since the discovery of innate immune system more than 100 yr ago by Metchinikoff. Now, the importance of innate immunity is being recognized by most immunologists which is reflected by the Nobel Prize in Physiology or Medicine bestowed on the discoverers of the innate immune receptors in 2011. Most importantly, full-strength specific immunity is not generated without participation of innate immunity, as Dr. Charles Janeway explained as the 'immunologist's dirty little secret' long before the discovery of innate immune receptors (1).
My interest in the role of innate immunity related to the pathogenesis of T1D was started by the results from my research group regarding pancreatic β-cell apoptosis in T1D. Apoptosis of pancreatic β-cells is the last step in the development of T1D in that after a long sequence of autoimmune processes, finally β-cell apoptosis occurs leading to critically reduced β-cell mass and clinical T1D (2, 3). Then, what is the initial event that is more important both scientifically and clinically? Clues came from the findings that transient wave of physiological β-cell apoptosis occurs with a peak at 2-3 weeks of age during the pancreas development (4, 5) in nonobese diabetic (NOD) mouse, a classical T1D model. This event coincides with the onset of autoimmunity in those mice, which was reflected by the proliferation of naïve diabetogenic T cells specifically in the pancreatic lymph nodes of NOD mice at 3 weeks of age (6). While apoptotic cells are usually rapidly engulfed by macrophages leaving no inflammation, apoptotic cells may undergo secondary necrosis when not removed in time by macrophages, which may trigger innate immune responses. Such events may occur in NOD mice that have defects in phagocytic capability (7). Indeed, it was observed that, when left untreated for more than 36 hr, apoptotic insulinoma cells undergo secondary necrosis characterized by propidium iodide uptake due to permeabilization of plasma membrane, which can stimulate antigen-presenting cells such as macrophages through Toll-like receptor 2 (TLR2), an innate immune receptor, inducing release of inflammatory cytokines including TNF-α and IL-12 (8). We also found that priming of diabetogenic T cells by dendtiric cells due to β-cell death in vivo occurring in the pancreatic lymph nodes through TLR2. Furthermore, the incidence of T1D after multiple low-dose streptozotocin administration was significantly decreased in TLR2-knockout mice, and that of spontaneous T1D was remarkably reduced in TLR2-knock NOD mice that were generated by backcrossing TLR2-knockout mice to NOD mice for more than 11 generations (8), clearly indicating the role of TLR2 in the development of T1D (Fig. 1). Role of other TLRs such as TLR9 in T1D has also been suggested in a report showing that activation of TLR9 of plasmacytoid dendritic cells by β-cell apoptosis (9). Recent investigation by others showed significant reduction of the incidence of T1D in NOD mice with knockout of TLR2 or MyD88 (10), a common adaptor for most TLR, but not with other TLRs (Alan Baxter, Immunology of Diabetes Society 13th International Congress, Lorne, Australia, 2013). No decrease in T1D incidence in TLR6- or TLR1-knockout NOD mice suggests possible involvement of TLR2 homodimers rather than classical TLR2-TLR6 or TLR2-TLR1 heterodimers in the recognition of death-associated molecular pattern (DAMP) from apoptotic β-cells.
Inhibition/treatment of T1D through tolerization of innate immune receptors
Since these results suggested the possibility that TLR2 blockade could inhibit T1D, we investigated whether TLR2 tolerization with chronic administration with TLR2 agonists could inhibit or treat T1D. Indeed, chronic treatment with Pam3CSK4 induced TLR2 tolerance in vitro and in vivo, which was shown by the attenuation of diabetogenic T cell priming by dendritic cells in pancreatic lymph nodes. Furthermore, the development of T1D in NOD mice was significantly inhibited by prolonged administration of 100 µg Pam3CSK4 3 times a weeks for 3 weeks since 3 weeks of age (11). TLR2 tolerization also could treat established T1D in conjunction with islet transplantation that restores already critically reduced β-cell mass. Since the supply of pancreatic islets is limited in clinics, we next employed another strategy restoring reduced β-cell mass - dipeptidyl peptidase 4 (DPP4) inhibitors that have positive effects on β-cell survival and mass. Indeed, the combination of TLR2 tolerization and DPP4 inhibition could reverse newly established T1D of NOD mice in more than 80% (12). These results show that innate immune receptors such as TLR2 play a crucial role in the initiation and perpetuation of T1D, and that understanding of the role of innate immunity could be a key to the inhibition or treatment of T1D (13).
ROLE OF INNATE IMMUNITY IN T2D
NLRP3 as a sensor of lipid injury
T2D has been traditionally regarded as a purely metabolic disorder. However, recent investigation has revealed the role of chronic inflammation in the pathogenesis of T2D (14, 15). Lipid such as palmitic acid has been considered to act on immune receptors such as TLR4 (16, 17), which induces chronic low-grade tissue inflammation. Besides lipids, microbiota in the intestine can have impact on mucosal innate immunity, which will affect local and systemic inflammation, immunity and host metabolism. We recently have reported that recently identified anaerobic bacteria called Akkermansia can improve metabolic profile and systemic inflammatory tone after high-fat diet (18).
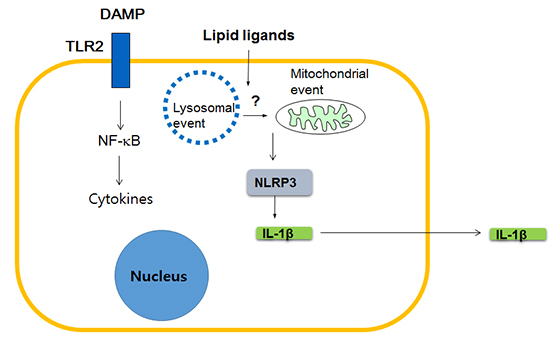
Which innate immune receptors then are critical for T2D and metabolic syndrome? While diverse innate immune receptors such as TLR4 or TLR2 have been previously claimed to be important in the development of obesity-induced diabetes (19), a couple of recent papers showed a possible critical role of NOD-like receptor family, pyrin domain containing 3 (NLRP3). NLRP3 is a member of NLRP subfamily of Nod-like receptor (NLR) family that, as a constituent of inflammasome complex, plays a crucial role in the maturation and release of IL-1β (20). These papers showed a positive correlation between pro-IL-1β or NLRP3 mRNA expression and body weight, and also improved glucose tolerance in NLRP3-knockout obese mice. Release of IL-1β in response to palmitic acid in combination with LPS was also demonstrated in vitro, together with the evidence indicating the involvement of NLRP3 in such in vitro IL-1β release (21). Subsequent works by other investigators showed potential role of the inhibition of NLRP3-mediated inflammasome activation in the therapeutic effects of antidiabetic medicines such as glyburide or metformin (22, 23), while the detailed molecular and cellular mechanisms in such events are not clearly elucidated (24). Regarding the molecular mechanism of NLRP3 activation, NLRP3 self-oligomerization after contact with DAMP or PAMP (pathogen-associated molecular pattern), subsequent binding to an adaptor protein called apoptosis-associated speck-like protein containing a CARD (ASC) and cleavage of procaspase-1 leading to the formation of active caspase-1 and maturation of IL-1β from pro-IL-1β, are well characterized (25). However, steps upstream of NLRP3 oligomerization are not entirely clear, while lysosomal and mitochondrial events appear to be involved (26, 27) (Fig. 2)
So far, I discussed about the role of NLR such as NLRP3 linking lipid injury to low-grade tissue inflammation, however, possible role of other innate immune receptors should also be considered. While it is still controversial whether lipid such as palmitic acid can directly activate TLR such as TLR4 (16, 17), a recent paper showed that palmitic acid bound to fetuin A, a glycoprotein that is released from the liver and can stimulate cytokine release from macrophages, can activate TLR4 (28). Furthermore, TLR4 stimulation is believed to be essential for maximal inflammasome stimulation since pro-IL-1β mRNA expression should be enhanced by stimulation of non-NLR innate immune receptors such as TLR4 before cleavage and maturation of caspase-1 and pro-IL-1β protein (25). Thus, collaboration between NLRP3 and other innate immune receptors such as TLR4 appears to be necessary for sufficient inflammasome activation and IL-1β release (Fig. 2).
Inflammasome activation in human-type diabetes
Possible role of NLRP3 in human T2D also has been suggested in experiments showing the capability of human-type islet amyloid polypeptide (hIAPP) to activate NLRP3 inflammasome (29), which could be mechanistically similar to the NLRP3 activation by fibrillar Aβ oligomer (30). hIAPP oligomers accumulating in islets of human T2D patients but not in those of rodent T2D, have similar conformation to that of Aβ oligomer, suggesting a common pathogenetic mechanism between human diabetes and Alzheimer's disease (31). Thus, NLRP3 activation by amyloidogenic hIAPP but not by non-amyloidogenic rodent IAPP suggests more important role of NLRP3 and inflammasome activation in the pathogenesis of human T2D compared to rodent T2D. These results also imply that NLRP3 activation is involved not only in insulin resistance of T2D but also in β-cell dysfunction of T2D, and may explain a dominant role of improved β-cell function rather than improved insulin sensitivity in the amelioration of T2D after administration of Anakinra, an IL-1 receptor antagonist, to T2D patients (32).
These results suggest that pharmacological manipulation of NLRP3 inflammasome activation may be a new modality for the treatment of metabolic syndrome or T2D, particularly human T2D, associated with lipid injury.
CONCLUSION
As discussed above, innate immunity is important in the pathogenesis of not only autoimmune T1D but also in T2D. Sine specific innate immune receptors involved in T1D and T2D are being elucidated, immunotherapy targeting specific innate immune receptors among a large number of innate immune receptors will be available for these disorders due to the intensive investigations by motivated immunologists and diabetologists. Such information and discovery may also be useful for treatment of other diseases such as obesity, metabolic syndrome, atherosclerosis or gout.
 XML Download
XML Download