

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Glycogen storage disease type V (GSD-V), also known as McArdle's disease, is the most common disorder of muscle glycogenosis (1). GSD-V is caused by alterations in the PYGM which encodes myophosphorylase. Myophosphorylase catalyzes the phosphorolytic cleavage of glycogen to glucose-1-phosphate in skeletal muscle. Therefore, enzyme deficiency results in the defective breakdown of glycogen into glucose and glycogen accumulation in muscle (2).
GSD-V is inherited in an autosomal recessive pattern, and the prevalence has been estimated at about 1 in 100,000-167,000 (3, 4). Most adult patients present with typical clinical features such as exercise intolerance, episodic myoglobulinuria and the second-wind phenomenon. In addition, the resting serum creatine kinase (CK) level, forearm exercise test, and electrophysiological studies can contribute to diagnosis. Subsequent mutation analysis and myophosphorylase activity assay can be used to confirm the diagnosis.
There has only been one case report of Korean patients with GSD-V (5). However, the previous report did not feature typical clinical presentations of GSD-V such as myoglobulinuria and the second-wind phenomenon. Here, we report a Korean case of GSD-V with typical clinical and laboratory findings.
CASE DESCRIPTION
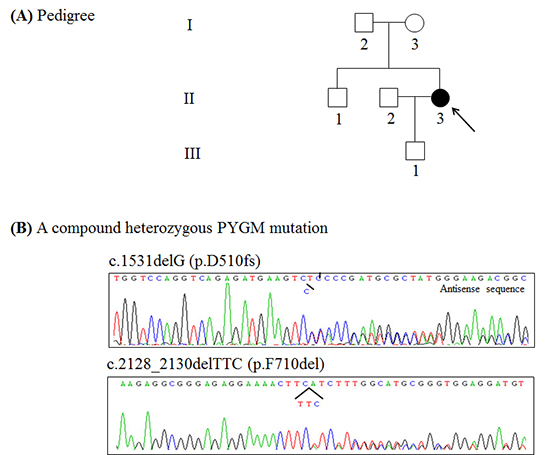
A 32-yr-old woman (Fig. 1A, II-3) was referred to our clinic due to easy fatigability, muscle cramps and myoglobulinuria in June 2013. Her family history was unremarkable. She recalled easy fatigability, muscle cramps and contractures after intense exercise during physical education classes since early adolescence. In addition, she had several episodes of dark urine following intense exercise such as long-distance running. These symptoms of exercise intolerance had been aggravated one year before. She also reported marked improvement in exercise tolerance after about 20 min, a process termed the 'second-wind phenomenon'; specifically, she had seen improvement in excessive fatigue, breathlessness, and tachycardia. Physical examination did not show fixed muscle weakness and muscle atrophy. All of her sensory modalities and tendon reflexes were intact. Laboratory studies revealed a serum creatine kinase (CK) level of 1,161 IU/L (normal<215 IU/L) at rest. Nerve conduction study (NCS) and needle electromyography study (EMG) did not show any abnormalities at rest. The non-ischemic forearm exercise test showed no increase in venous lactate level with a normal increase in ammonia (Fig. 2). To confirm easy fatigability and weakness after exercise, we performed motor NCS on the ulnar nerve after voluntary maximal contraction for two minutes and 50 Hz nerve stimulations for one second. However, the amplitudes of the compound muscle action potentials did not decrease after maximal contraction and nerve stimulation. In addition, EMG did not show silent electrical activities during muscle cramps. Based on the clinical and laboratory features, she was diagnosed with glycogenoses with exercise-induced weakness, especially GSD-V. Therefore, we performed PYGM genotyping.
Mutational analysis
A DNA sample was extracted from peripheral leukocytes using the Quick Gene blood kit (Fujifilm, Tokyo, Japan). The primer set that covers all 20 exons and their flanking intron regions of the PYGM was designed with Primer 3web v.4.0 (http://primer3.wi.mit.edu/) using sequences from the Gen Bank accession number NT_167190.1. All the exons of the PYGM were sequenced using BigDye Terminator v.3.1 (Applied Biosystems, Foster City, CA, USA) followed by polymerase chain reaction. Electrophoresis and analysis of the reaction mixtures were done with ABI3130xl Genetic Analyzer (Applied Biosystems). A compound heterozygous mutation was identified; one allele had a frameshift mutation of c.1531delG (p.D510fs) in exon 13 and the other allele had a deletion mutation of c.2128_2130delTTC (p.F710del) in exon 17 (Fig. 1B). These mutations have been previously reported to be underlying causes of GSD-V in Japanese patients (6).
DISCUSSION
We described a Korean patient with PYGM mutations who had typical clinical and laboratory findings for identification of GSD-V. Even though GSD-V is the most common disorder of muscle glycogenosis, our patient is only the second case of genetically proven GSD-V in Korea.
Four characteristic clinical features are important for initial suspicion of GSD-V. First, exercise intolerance such as easy fatigability, muscle cramps and contractures is triggered by static contractions and dynamic exercise. Almost all patients are admitted to the hospital for exercise intolerance. The pathophysiology of exercise intolerance is not fully understood, but may be associated with impaired glycolysis (2). A deficient glycogen-dependent adenosine triphosphate (ATP) supply might result in down-regulation of Na+/K+ pumps in myocytes, leading to loss of membrane excitability (7, 8). The defective breakdown of glycogen into glucose is reproduced by the forearm exercise test. The forearm exercise test showed no increase in lactate level with a normal increase in the ammonia level. The forearm exercise test was originally carried out under ischemic conditions; however, we did not conduct the test under ischemic conditions because the diagnostic value is just as good in non-ischemic conditions and muscle injury can be avoided (9). In addition, several previous studies demonstrated loss of membrane excitability as the amplitude of CMAPs decreased after maximal contracture and high frequency repetitive nerve stimulation, and silent electrical activities during muscle cramping (10, 11). However, we did not identify these findings in the patient presented in this case study. It may be associated with benign clinical symptoms without fixed muscle weakness.
Second, the serum CK level is elevated in almost all patients with GSD-V even in the absence of exercise. One recent report indicated that hyperCKemia (>200 IU/L) was present in 99% of patients and the majority (79%) of patients had a level greater than 1,000 IU/L (4). The elevation of serum CK level at rest is likely associated with muscle damage due to impaired glycolysis during exercise. However, the serum CK level at rest did not correlate with clinical severity because exercise-induced muscle damage can stimulate muscle repair and adaptive hypertrophy (12). In addition, one study demonstrated no significant difference in serum CK level at rest between physically active and inactive patients (4).
Third, about half of all patients with GSD-V have experienced one or more episodes of hyperCKemia (several thousand IU/L) or myoglobulinuria after intense exercise. In addition, about 50% of patients with myoglobulinuria develop acute renal failure, which is almost always reversible, but nevertheless requires emergency treatment (13).
Fourth, the second-wind phenomenon, an especially unique feature of GSD-V, differentiates GSD-V from other muscle glycogenoses such as muscle phosphofructokinase deficiency (13). This phenomenon is reported by almost patients during the medical interview. If it is not found in the medical history, it is easily reproduced by diagnostic cycle test (14). Therefore, it is the key finding that leads to initial suspicion of GSD-V. This phenomenon is caused by an increased supply of glucose and free fatty acids after a long period of exercise. These changes lead to a switch in metabolic pathways from endogenous glycogenolysis to oxidative phosphorylation of blood-borne fatty acids (15). However, continuing to exercise also results in severe muscle cramps and myoglobulinuria.
Besides these major clinical features, some patients with GSD-V can also show fixed muscle weakness. Most patients including the patient in this report did not complain of muscle weakness, but approximately one-third of patients develop proximal muscle weakness with aging (2, 16).
Mutation analysis and the myophosphorylase activity assay are used to confirm the diagnosis of GSD-V; however, molecular analysis is more important and more commonly used because muscle biopsy is needed for the enzyme activity assay. At last count in February 2013, 131 different mutations have been reported in the human gene mutation database (http://www.hgmd.org). Genotype has not been correlated with the severity of clinical phenotype in previous studies (17, 18). However, there has been a higher incidence of some genotypes in specific ethnic groups; for example, p.R50X and p.G205S in the white population, p.F710del in the Japanese population, and p.W798R in the Spanish population (13). Likewise, two mutations in our patient have previously been reported in Japan, a neighboring country of Korea (6). In particular, the p.F710del mutation has been identified in 73% of Japanese patients (19).
In conclusion, we identified a compound heterozygous mutation of the PYGM in a Korean patient with typical clinical and laboratory findings. We recognize that detailed clinical and laboratory analysis is only the first step in the diagnosis of disease, especially GSD-V.


 XML Download
XML Download