

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Timothy syndrome (TS) is also known as long QT syndrome (LQTS) type 8. TS is a multisystem disorder characterized by cardiac, hand, facial, and neurodevelopmental disorders and especially associated with syndactyly. It is a genetic disorder caused by mutation in the Cav1.2 L-type calcium channel gene (CACNA1C). This mutation is related to arrhythmia, including bradycardia, atrioventricular block, torsades de pointes ventricular tachycardia, and ventricular fibrillation. TS is a rare disease, with only 25 molecularly confirmed cases reported to date since an L-type calcium channel mutation was first described in this syndrome (1-3) and there was no genetically proven case in Asia.
TS is often regarded as lethal disease, and most common cause of the reported deaths in patients with TS is ventricular tachyarrhythmia at the mean age of 2.5 yr (2). We report the case of a patient who initially presented with cardiac arrest, which was diagnosed as TS by a gene study.
CASE DESCRIPTION
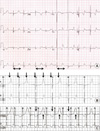
A 30-month-old boy suddenly experienced cardiac arrest during anesthesia induction with sevoflurane and nitric oxide before undergoing plastic surgery for bilateral cutaneous syndactyly of the fingers (third, fourth, and fifth digits) on June 7, 2004 (Fig. 1). After 10 min of resuscitation, sinus rhythm was recovered, and an electrocardiogram (ECG) showed a prolonged QT interval (QTc, 0.58-0.60 sec). Before the operation, the patient had never undergone ECG. During the Holter study, T-wave alternans and functional 2:1 AV block due to QT prolongation was also observed, suggesting LQTS (Fig. 2).
On the patient's echocardiogram, intracardiac anomaly was absent, and ventricular function was normal. The preoperative biochemical analysis result was normal; in addition, the electrolyte level was normal. The patient had a round face, small upper jaw and teeth, and nearly bald head; he had fused fingers but normal toes, no history of seizure or syncope, and normal neurodevelopment. The patient was born via vaginal delivery without any complications. The patient's family, particularly his older sister, had no history of syncope, seizure, or sudden death. The QT intervals of the father, mother, and sister were not significantly prolonged, as determined by ECG. The epinephrine stress test result of his sister showed a paradoxical QT interval prolongation (QTc baseline 0.4 → peak 0.53 sec), but she was asymptomatic. Genetic tests were not administered to the patient's parents and sister. No other arrhythmic events were detected, and a β-blocker (propranolol) was administered 3 times a day (1 mg/kg/dose) to prevent further tachycardia and collapse event. After supportive care for hypoxic brain damage, the patient was discharged with propranolol (2 mg/kg/day), and his neurological function returned to its premorbid state after one month. Although there was persistent severe QTc prolongation with intermittent 2:1 functional AV block, the patient underwent repeated plastic surgery for syndactyly without any arrhythmic event (at the age 4 and 5 yr), with propranolol and mexiletine. Mexiletine was prescribed from the ages of 4-7 yr on the supposition that he might be LQTS type 3 due to very long QTc interval, late appearing high amplitude T wave, and repolarization-dependent atrioventricular block. It has been discontinued since the result of SCN5A genes mutation test came out negative informally. The short acting β-blocker, propranolol, was changed to long acting β-blocker, atenolol, at the age of 10 yr old.
At the age of 6 yr, one morning, the patient had a generalized tonic-clonic seizure that lasted for 10 min; his blood glucose level was 38 mg/dL, with no other symptoms. There were no specific findings on his electroencephalogram and magnetic resonance imaging scan, and no recurring seizure was reported. Moreover, the result of the gene study performed during this time was negative for KCNQ1 and KCNH2 mutations. At the age of 10 yr (2011), mutations were absent in the KCNE1, KCNE2, and SCN5A genes, but point mutation c.1216G > A in exon 9 in CACNA1C was confirmed (Fig. 3). However, the patient's family refused to undergo the recommended gene study.
The patient was administered a β-blocker (atenolol at 0.35 mg/kg/dose bid) for 8 yr. The QT prolongation on ECG (QTc, 0.65 sec) and T-wave alternans on Holter monitoring persisted, but syncope or cardiac arrest did not recur during the 8-yr follow-up period.
DISCUSSION
TS is caused by p.Gly406Arg mutation in the CACNA1C. This mutation reduces Cav1.2 L-type calcium channel inactivation, which maintains depolarizing Ca2+ currents during the plateau phase of the cardiac action potential, and consequently leads to prolonged myocardial action potential and QT prolongation. Prolonged ventricular repolarization and refractory period could cause 2:1 AV block (2, 4), putting patients with TS at risk of life-threatening ventricular arrhythmia (4, 5). Bradycardia and 2:1 AV block are sometimes associated with fetal distress during the prenatal period (6). Approximately 70% of patients with TS have congenital heart defects, including patent ductus arteriosus, ventricular septal defect, tetralogy of Fallot, and hypertrophic cardiomyopathy (1, 3, 7).
Cutaneous syndactyly (in fingers or toes), distinct craniofacial features, and neuropsychiatric involvement (developmental delay and autism) are extracardiac manifestations of TS. Cutaneous syndactyly also can be seen in LQTS type 7 (Andersen-Tawil syndrome), but mostly in toes. So other features of the disease including periodic paralysis will be helpful to diagnose. Patients with TS have low-set ears, flat nasal bridge, small upper jaw, and small misplaced teeth and are bald from birth until the first 2 yr of life, when thin scalp hairs begin to grow (1). Given that the Cav1.2 L-type calcium channel is widely expressed in multiple adult and fetal tissues, including those of the gastrointestinal system, brain, lungs, immune system, and testis, the occurrence of facial dysmorphism, myopia, immune deficiency with recurrent infections, intermittent hypoglycemia, and hypothermia as well as developmental delay or autism are common (2, 5, 7).
Although TS is inherited in an autosomal-dominant manner, it usually results from a de novo mutation. Thus, the risk to a proband's sibling(s) is low. However, because of parental germline mosaicism, the sibling(s) of a proband may be at increased risk of inheriting a CACNA1C mutation (1, 8). Generally, the diagnosis of TS is possible within the first few days of life owing to markedly prolonged QT interval, bradycardia, and 2:1 AV block (1, 3); hence, delayed diagnosis until the ages of 2 to 4 yr, as in the present case, is rare. TS is diagnosed by clinical features, including long QT interval on ECG, and molecular genetic analysis. As described earlier, in the present case, the patient had typical findings for TS, which are prolonged QT interval, T-wave alternans, functional 2:1 AV block, syndactyly, and seizure related to hypoglycemia. Cardiac arrest during anesthesia is also possible in LQTS because anesthesia triggers arrhythmia. It has been reported that arrhythmia during anesthesia in LQTS patients often occurs, and especially polymorphic ventricular tachycardia known as torsades de ponites holds a large majority. It potentially affects repolarization of cardiac myocyte and augments sympathetic tone with insufficient anesthesia, hypertension, bradycardia, tachycardia, hypothermia, hypoxemia, and hypocapnia or hypercapnia (9). In addition a number of medications during anesthesia can bring about QT interval prolongation. In this case, he was anesthetized by sevoflurane, thiopental, atracurium and suffered cardiac arrest induced by arrhythmia within a few minutes. Those medications were relatively safe and uncontentious in LQTS patients. It was difficult to find another factor of arrhythmia except anesthetic process. Therefore, careful cardiac monitoring is required during anesthesia (10, 11).
Ventricular tachyarrhythmia is the leading cause of death in 80% of the patients with TS (12). Because TS is rare, precise LQTS subtype-specific management has not been established. Usually β-blockers, which maintain QT interval stability, and other antiarrhythmic drugs (verapamil or mexiletine) are prescribed to prevent further ventricular arrhythmia. Despite medical management, lethal ventricular arrhythmia can still occur; hence, left cervical sympathetic denervation or implantable cardioverter defibrillator should be considered to prevent death from sudden cardiac arrest (8, 13).
In conclusion, the present case is that of genetically proven TS, confirmed by the mutation p.Gly406Arg in CACNA1C, which was incidentally discovered when the patient underwent a genetic study after a sudden cardiac arrest during anesthesia. To our knowledge, this is the first report of a case of genetically proven TS in an Asian patient. The co-occurrence of LQTS and syndactyly is a rare but distinguishing finding in TS. TS should be suspected due to fatal ventricular arrhythmia. Therefore, we emphasize that all syndactyly patients should undergo careful ECG monitoring.


 XML Download
XML Download