

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Sheldon-Hall syndrome (SHS, MIM #601680), or distal arthrogryposis (DA) type 2B, is a rare autosomal dominant, inherited arthrogryposis syndrome characterized by congenital contractures of two or more different body areas, with no primary neurological disease. The common clinical features of SHS include contractures of the distal limb joints causing camptodactyly, and clubfeet, triangular face, downslanting palpebral fissures, prominent nasolabial folds, small mouth, and high arched palate (1). SHS is distinguished as a separate entity from Freeman-Sheldon syndrome (FSS, DA type 2A, MIM #193700), another DA type 2 syndrome, because FSS includes striking contractures of the orofacial muscles and is a more severe DA than SHS (2).
DA is a genetically heterogeneous disorder, with at least 10 different DA types characterized to date (3). Although the prevalence of arthrogryposis is known to be 1/3,000 (4), there are no epidemiological data for DA and fewer than 100 cases of SHS have been reported in the literature (5). To date, five genes that encode the skeletal muscle contractile fiber complex have been confirmed as the causative genes of DA types 1 and 2. Among these, mutations in TNNI2, TNNT3, TPM2, and MYH3 have been shown to cause SHS (6).
In this study, we present for the first time a Korean family with two generations of SHS, resulting from a rare TPM2 mutation, who manifested the facial features, short stature, and DA typical of SHS.
CASE DESCRIPTION
We studied a family in which SHS affected two individuals, a mother and her daughter. A one-month-old Korean girl was referred to the Seoul National University Children's Hospital, for the evaluation of multiple congenital contractures of both hands and feet on December 22th, 2011. She was the first baby in the family. The patient was delivered by cesarean section, because of her mother's narrow pelvic bones, at the 38th week of gestation; her birth weight was 2.56 kg (3rd-10th percentile) and her birth length was 43.0 cm (< 3rd percentile). On prenatal ultrasonography, bilateral clenched hands and bilateral talipes equinovarus were suspected.
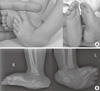
At the initial examination after birth, camptodactyly, overlapping fingers, and adducted thumbs were identified in both her hands. A calcaneovalgus deformity with congenital vertical talus in the right foot and an equinovarus deformity in the left foot were observed (Fig. 1). She also displayed subtle facial dysmorphism, including a triangular face, downslanting palpebral fissures, and a small mouth. Ten days after discharge from the neonatal unit of our hospital, the infant was admitted to a regional hospital for pneumonia, presumed to be aspiration pneumonia, for one week.
At the age of one month, the patient's weight was 2.7 kg. She was on frequent bottle feeding with small amounts of whole milk, because her micrognathia and small mouth caused feeding difficulties. Physical therapy for her multiple camptodactyly of the fingers was commenced at that time. One month later, she suddenly showed difficulty breathing one hour after feeding at home and was transferred to the emergency room of a regional hospital. Although cardiopulmonary resuscitation was applied on arrival at the emergency room, she did not recover and died. Aspiration pneumonia was considered to be the cause of her death.
The mother was 25 yr old. Her parents and two siblings were healthy. She was born in the 34th week of gestation with a birth weight of 1.8 kg (25th-50th percentile). She had difficulty breast-feeding because of her retrognathia and small mouth, so she was bottle fed. She began walking independently at the age of 19 months. Her height was 153 cm (5th-10th percentile) and her weight was 53 kg (25th-50th percentile). Her facial characteristics include a triangular face with downslanting palpebral fissures, low set ears with attached earlobes, small mouth, high arched palate, receding chin, prominent nasolabial folds, broad and long nasal bridge and root, and long philtrum (Fig. 2). A short neck and sloping shoulders were also noted. She was of normal intelligence, but mild bilateral sensorineural hearing impairment was detected with pure-tone audiometry.
The mother had multiple congenital contractures of the distal limbs. This was accompanied by camptodactyly with ulnar deviation of all 10 fingers on both hands, and bilateral talocalcaneal coalition, with left foot clubbing. Contracture release operations of fingers had been performed five times, and the talocalcaneal coalition had been excised. The contractures of her fingers were greatly improved, but persisted to some extent. Dorsiflexion of both feet was impossible. She also had mild contractures of both knees. However, she did not complain of gait disturbance and the motion in neither hip joint was limited.
Familial SHS was suspected based on the family's medical history and the findings and the findings of physical examinations. A direct sequencing analysis of the MYH3, TNNI2, TNNT3, and TPM2 genes was performed to confirm SHS at the molecular genetic level. A previously reported mutation in exon 4 of the TPM2 gene, c.397C > T (p.R133W), was detected in the daughter (Fig. 3). A genetic analysis of her parents revealed that the mother carried the same mutant allele. Familial SHS was thus confirmed in the mother and daughter. No mutation was identified in MYH3, TNNI2, or TNNT3. The TPM2 gene is also known to be one of the causative genes of nemaline myopathy. Therefore, we examined the mother with electromyography (EMG), although there was no subjective muscle weakness in her daily activities. The EMG findings revealed generalized myopathy, with relative sparing of the slow-twitch muscle fibers. We provided genetic counseling for the family, and the mother is planning a prenatal genetic diagnosis in her next pregnancy.
DISCUSSION
DA is clinically defined as a group of inherited limb malformation syndromes, with contractures primarily involving the distal limbs, and skeletal muscle weakness (7). DA shows marked clinical and genetic heterogeneity, with at least 10 different forms of DA documented to date (3), The classification of the different types of DA syndromes is difficult because of the reduced penetrance and variable expression of the disease. FSS and SHS are the most distinctive DA subtypes because they include additional facial characteristics.
Recently, mutations in the genes encoding the skeletal muscle contractile fiber complex (TNNI2, TNNT3, TPM2, MYH3, and MYBPC1) have been identified as the causes of DA types 1 and 2 (6). Among these genes, mutated MYH3 is thought to be the most common cause of SHS, with MYH3 mutations found to account for 32% of SHS (8). We screened the MYH3 gene first, but found no mutation. Therefore, we sequenced the TNNI2, TNNT3, and TPM2 genes directly and identified the p.R133W mutation in TPM2.
Tropomyosin is central to the control of calcium-regulated striated muscle contractions via its interaction with actin and the troponin complex (9). Tropomyosin has three isoforms, α, β, and γ-tropomyosins, and β-tropomyosin is encoded by the TPM2 gene. TPM2 is mainly expressed in slow-twitch muscle fibers, but β-tropomyosin protein expression is higher in fast-twitch muscle fibers than in slow-twitch fibers (10). Different TPM2 mutations have been identified in association with a wide range of skeletal myopathies besides DAs, including congenital myopathy, nemaline myopathy, and cap disease (11, 12). However, DAs associated with TPM2 mutations are quite rare: only two TPM2 mutations have been reported as causes of DA, each in only one family. The p.R91G mutation was found in one family with DA type 1 (13), and the p.R133W was identified in one family with SHS with muscle weakness. However, the muscle weakness associated with this mutation was not accompanied by progressive muscle wasting or histopathological abnormalities, except slow-twitch fiber predominance (14).
Although we did not take a muscle biopsy and our patient did not complain of motor weakness during her daily activities, her EMG results suggested generalized myopathy, with relative sparing of slow-twitch muscle fibers. The previously reported patients with the p.R133W mutation were 28 and 65 yr old at the time, and had prominent muscle weakness, mainly in the hands and feet. Mild progression of this muscle weakness was described only by the 65-yr-old patient. Our patient is 26 yr old now, and further long-term follow-up with regular monitoring of her motor function will be necessary.
The mechanism by which certain mutations in TPM2 cause multiple congenital contractures is still unclear. One hypothesis is that mutations in the tropomyosin genes cause changes in the actin-myosin interaction and modify the contractile speed (11). A previous in vitro study of the p.R133W mutant tropomyosin also showed that this mutation caused a slower actin-myosin attachment rate and a faster detachment rate (15, 16). Hence, this mutant protein may cause a reduction in the number of myosin molecules in the strong actin-binding state, resulting in overall muscle weakness and DA (15, 17).
Here, we have reported a family with SHS resulting from a TPM2 mutation, who showed typical clinical phenotypes. Although SHS is a rare disease, it can be transmitted as an autosomal dominant condition and familial recurrence has been reported for approximately 50% of cases (18). Progressive motor weakness is also possible, and associated feeding problems may lead to a sudden deterioration of the clinical condition, as in our infant patient. An accurate diagnosis followed by appropriate management and genetic counseling should be provided to these patients.


 XML Download
XML Download