

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Fluid accumulation in cardiac interstitium has been associated with many diseases or clinical states and results in cardiac dysfunction (1). Myocardial infarction (MI) induces myocardial edema with expansion of both interstitial and intracellular compartments (2). During the ischemia, interstitial edema develops due to the leaky myocardial capillaries, and is followed by swelling and dysfunction of cardiac myocytes (3). The resulting impairment of systolic and diastolic functions is of prognostic importance in the patient survival after myocardial infarction (4, 5).
Aquaporins (AQPs) are a family of small integral membrane proteins that primarily transport water across the cell membrane along osmotic gradients. Until now, 13 AQPs have been found in mammals (AQP0-12), some of which permit transcellular passage of glycerol and urea as well as water (AQP3, 7, 9, and 10) (6, 7). AQPs have been demonstrated to play a pivotal role in physiology and pathophysiology of body water balance (8). Experiments using AQP4-null mice revealed that AQP4 deletion protects from cellular (cytotoxic) brain edema produced by brain ischemia, but rather aggravates vasogenic (fluid leak) brain edema produced by brain tumor, providing strong evidence for AQP4 involvement in cerebral water balance (9). Based on the findings in brain edema, the implication of AQPs in myocardial water balance might also be postulated, but has not been determined.
In addition, the expression of AQP subtypes and their precise localization in myocardial tissue still remain unclear. In the mouse heart, the presence of AQP1, 4, 6, 7, 8, and 11 mRNA has so far been described, whereas only the presence of AQP1 and AQP4 was confirmed at the protein level (10, 11). However, for other AQPs, different results have been reported depending on the species and techniques used (12-14).
In this study, we characterized the expression patterns of AQP subtypes in normal mouse heart and investigated whether their expression levels change during the development and reabsorption of myocardial edema after MI.
MATERIALS AND METHODS
Animal preparation
All the procedures were approved by Dong-A University Medical School Institutional Animal Care and Use Committee (DIACUC-10-6-1). Total of 18 CD1 mice, 10-12 weeks of age, were used and divided into three groups; sham surgery group (Control, n = 6), 1-week MI group (MI-1W, n = 6) and 4-week MI group (MI-4W, n = 6). All the animals were anesthetized by intraperitoneal (IP) injection of 3.5% chloral hydrate (10 µL/g), endotracheally intubated, and mechanically ventilated using the Inspira-Advanced Safety Ventilator (ASV, NP55-7050, Harvard Appartus, Holliston, MA, USA), which supplied 0.25-0.30 mL room air 130 times per minute. The animal was moved onto its right side, and then the heart was exposed via a left thoracotomy. After removing the pericardium, the left anterior descending artery was visualized using a stereomicroscope (Nissho Optical, Kodama-gun, Japan), and occluded with a 9.0 nylon suture (Tyco Healthcare, North Haven, CT, USA). The suture was placed 0.3 mm distal to the atrioventricular junction. Occlusion was confirmed by observing the left ventricular pallor immediately after ligation. The chest was closed, the lungs reinflated, and the animal was moved into the prone position until spontaneous breathing occurred.
Measurement of cardiac function
Echocardiography analysis was performed baseline and each endpoint time using an echocardiography system with a Sonos 4500 and a 15-16 MHz transducer (Philips Corporation, Eindhoven, The Netherlands). The animals were sedated with anesthetic (3.5% chloral hydrate 1 mL/100 g, IP) and placed in the left lateral decubitus position. Parasternal long and short axis views were obtained in both M-mode and 2-dimensional echo images. Left ventricle end diastolic diameter (LVEDD) and left ventricle end systolic diameter (LVESD) were measured perpendicular to the long axis of the ventricle at the mid-chest level. Left ventricular ejection fraction (LVEF) was calculated automatically by the echocardiography system.
Tissue preparation
Animals were anesthetized and the thoracic cavity was opened to expose the heart. A 20-gauge needle connected with a micropump via a tube was inserted into the left ventricle, and the animal was slowly perfused with approximately 40-50 mL of phosphate buffered saline (PBS) until the fluid became clear. The hearts were cut into two equal transverse blocks from apex to base, either frozen in liquid nitrogen or fixed in 10% neutral buffered formalin and embedded in paraffin. Serial sections of 5-µm thickness were cut and used for further studies.
Assessment of myocardial water content
The hearts were removed at the different time points after MI and cut into two pieces just below the ligation suture. The lower half sections including the infarcted areas were weighed before (wet weight) and after drying to a constant weight in a desiccating oven at 65℃ (dry weight). The percent myocardial water content was calculated as: (wet weight-dry weight) × 100/wet weight.
Immunostaining
Frozen sections were air-dried, fixed in ice-cold acetone and blocked with 5% bovine serum albumin before immunostaining. Paraffin sections were deparaffinized with xylene, and serially rehydrated in 100%, 95%, and 70% ethanol. For epitope retrieval, slides were immersed into coplin jar containing citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) and boiled for 15 min. The jar was allowed to cool for 1 hr at room temperature (RT) and then slides were rinsed with washing buffer containing 0.025% Tween 20. After blocking for 2 hr, the sections were incubated with primary antibodies; rabbit polyclonal anti-AQP1 (1:300, Chemicon, Temecula, CA, USA), anti-AQP4 (1:300, Millipore, Billerica, MA, USA), anti-AQP6 (1:300, Chemicon), anti-AQP7 (1:300, Alpha Diagnostic, San Antonio, TX, USA) and anti-AQP9 (1:300, Alpha Diagnostic) overnight at 4℃. Secondary anti-rabbit horseradish peroxidase (HRP) linked IgG (1:100, Dako EnVision system, Glostrup, Denmark) was incubated for 1 hr at RT. The reaction was visualized with Dako DAB+ chromogen (1:100). Tissue sections were mounted, and then visualized using digital scanning microscope (Scanscope, Aperio, Vista, CA, USA).
Western blot analysis
Frozen tissue was homogenized in ice-cold homogenization buffer (300 mM sucrose, 20 mM Hepes [pH 7.4]) containing protease inhibitor cocktail (Roche, Basel, Switzerland) using a dounce homogenizer in ice. Nuclei and debris were removed after low speed centrifugation (1,000 g for 10 min) at 4℃ and the postnuclear supernatant was incubated with 2% Tween 20 for 1 hr in ice. The resultant crude membrane extract was loaded in each lane (50 µg per lane) to be separated by SDS-polyacrylamide gel electrophoresis (PAGE). After transferring the proteins onto a nitrocellulose membrane, the membrane was blocked with Tris-buffered saline-Tween (TBST; 20 mM Tris [pH 7.5], 145 mM NaCl, 0.05% Tween 20) containing 5% skim milk for 2 hr at RT, and then incubated with rabbit polyclonal anti-AQP1 (1:5,000), anti-AQP4 (1:5,000), anti-AQP6 (1:5,000) and anti-β-actin (1:2,500, Sigma-Aldrich, St. Louis, MO, USA), overnight at 4℃. After washing with TBST, the membranes were incubated with HRP-conjugated secondary antibodies (1:5,000, Cell Signaling, Danvers, MA, USA) for 45 min at RT and developed using a chemiluminescence detection system (Amersham Pharmacia Biotech, Little Chalfont, Bucks, UK).
RESULTS
AQP expression in normal heart
Immunostaining using antibodies against different AQP subtypes was conducted to determine which AQPs are expressed in normal heart. As shown in Fig. 1 and 2, AQP1, AQP4 and AQP6 proteins were abundantly expressed in normal cardiac tissue. However, neither AQP7 nor AQP9 were expressed in normal CD1 mouse heart (Fig. 1D, E, and 2D, E). AQP1 expressed in vascular endothelial cells lining blood vessels and red blood cells within them (Fig. 1A, 2A). Weak AQP1 staining was also observed in the cytoplasm of cardiac myocytes. Anti-AQP4 antibody revealed prominent labeling at the intercalated discs between cardiac myocytes, exclusively on the side of transverse region (Fig. 1B, 2B). No significant density of anti-AQP4 stain was detected within the cytoplasm of cardiac myocytes in normal heart. AQP6 expressed abundantly and distributed throughout the cytoplasm of cardiac myocytes in a striated pattern (Fig. 1C, 2C).
Functional and histological changes after myocardial infarction
Hematoxylin-eosin stain shows that the pale area of infarction was well-demarcated from the surrounding normal-appearing red area, and replaced with early granulation tissue after 1 week (Fig. 3A, MI-1W). Widened inter-fiber space was prominent at 1 week after MI presumably due to the interstitial fluid accumulation. Necrotic cardiac myocytes with disappeared nuclei and less prominent striation were observed in infarct area, whereas infiltration of polymorphonuclear leukocytes and proliferation of fibroblasts were found at the periphery of the infarcted region. Four weeks following MI, necrotic tissue was totally absorbed and replaced with fibrous scar tissue so that myofibroblasts became the major cell type in infracted myocardium (Fig. 3A, MI-4W). Cardiac dilatation with expanded ventricular lumen was noticeable after 1 week and became more prominent after 4 week with the progressive thinning of the left ventricular wall.
We analyzed time-dependent changes of cardiac size and function after MI using echocardiography. LVESD and LVEDD, which are the parameters of left ventricular diameter, were increased in MI-induced groups, either MI-1W or MI-4W, when compared with those in sham-operated control group (Fig. 3B). Both values increased time-dependently up to 4 weeks after MI. LVEF, the most commonly used measure of overall cardiac function, was maximally reduced at 1 week post-MI (46.1% ± 4.9% in MI-1W; 73.1% ± 5.1% in control, P < 0.01), then slightly recovered at 4 weeks post-MI (52.7% ± 6.0% in MI-4W, P < 0.05).
For the assessment of myocardial edema, percent myocardial water content was measured after MI. Myocardial water content increased by 8.7% at 1 week after MI and became slightly reduced at 4 weeks when compared with the control (Fig. 3C).
Changes of AQP expression after myocardial infarction
We next evaluated the MI-induced changes in expression patterns of AQP1, AQP4 and AQP6, which were confirmed to express in normal myocardium as shown in Fig. 1 and 2. The expression levels of all these AQPs increased following MI, not only at the periphery of infracted area but also in normal-appearing myocardium (Fig. 4). Although a slight up-regulation in cardiac myocytes was observed, MI-induced increase in AQP1 expression was mainly caused by increased signal from small blood vessels coursing between muscle fibers in non-infarcted areas up to 1 week (Fig. 4B). After 4 weeks of MI, much stronger staining intensity of AQP1 was observed inside cardiac myocytes (Fig. 4C). Intensity of AQP4 staining was also increased after MI so that intercalated discs looked more prominent (Fig. 4E). However, AQP4 staining pattern at intercalated discs appeared more undulated and widened at 1 week post-MI, presumably due to the interstitial edema. Interestingly, widely dispersed AQP4 staining became concentrated again at intercalated discs after 4 weeks (Fig. 4F). MI induced a dramatic up-regulation in AQP6 expression up to 4 weeks (Fig. 4H, I). Increased expression of AQP6 in cytoplasm made the sarcomeric striations of cardiac myocytes clearly discernible after MI. Individual sarcomeres as well as the space between them became widened after MI compared with those in sham operated control.
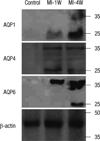
The MI-induced up-regulation of AQP expression was confirmed by western blot analysis. As shown in Fig. 5, regardless of the subtypes, AQPs showed increased expression after MI. However, the time courses of AQP expression following MI were different depending on the subtypes. Both AQP1 and AQP6 showed a stepwise increase in their expressions up to 4 weeks, whereas AQP4 reached peak expression at 1 week followed by a subsequent decline at 4 weeks.
DISCUSSION
The purpose of this study was to define the expression pattern of AQPs in normal heart and to investigate the changes in myocardial AQP expression after MI. We demonstrated here that AQP1, AQP4 and AQP6 are the main AQPs expressing in mouse heart and that the expressional level of these AQPs increases after MI with different time-dependent pattern, suggesting possible different roles for these AQPs on the pathogenesis of myocardial fluid accumulation accompanied by MI or on the post-infarction healing and remodeling process by facilitating myocardial fluid resorption.
The subtype-specific expression of AQPs and their precise localization in heart have not been fully determined. Although the multiple AQP transcripts have been identified in human and mouse heart by RT-PCR analysis or Northern blot, only the presence of AQP1 was demonstrated at the protein level in human heart and that of AQP1, AQP4, and AQP7 in mouse heart (10, 13, 15). Moreover, their precise localization in myocardial tissue using immunohistochemistry has not been reported or remains equivocal except AQP1 (11). Here, we confirmed not only the presence of AQP1, AQP4, and AQP6 but also their detailed localization in mouse myocardium using immunohistochemistry and western blot. In accordance to the findings of others, we also observed the expression of AQP1 at the sarcolemma of cardiac myocytes, although its level is weaker than that in endothelia lining the vasculature in heart (10, 15). However, as for AQP4 localization, we obtained different results than the previous researchers in that AQP4 is highly enriched at the intercalated discs, not the entire sarcolemma of cardiac myocytes as previously reported (10, 15). Whereas AQP4 localizes to the perivascular endfeet of astrocytes and skeletal muscle sarcolemma by binding syntrophin associated with the dystrophin-glycoprotein complex (17, 18), in cardiac tissue, AQP4 distributes exclusively at the intercalated discs. Recently, it has been reported that the main cardiac voltage-gated sodium channel, Nav1.5, is anchored to either the lateral membranes by binding syntrophin-dystrophin complex or the intercalated disc by binding synapse-associated protein 97 (SAP97) through PSD-95/Discs-large/ZO-1 (PDZ) domain binding motif in cardiac myocytes (19). Since AQP4 also has PDZ domain binding sequence at the C-terminal (20), binding of AQP4 to SAP97 might be one possibility for its localization to the intercalated disc.
Although the presence of AQP6 transcript was suggested previously by RT-PCR (10), we first observed that AQP6 proteins express in heart and localize inside the cardiac myocytes. AQP6 was originally identified as a homolog of AQP2 and expresses mainly in the intracellular vesicles of acid-secreting type A-intercalated cells of renal collecting ducts, colocalizing with H+-ATPase (21). AQP6 is permeable to anionic ions, especially nitrate, as well as water (22). AQP6 has been reported to express in synaptic vesicles and participate in their swelling with AQP1 (23). The possible roles of AQP6 in intracellular pH regulation, nitrate efflux or vesicular transport in cardiac myocytes remain to be established.
It is difficult to detect the myocardial edema-induced changes of ventricular wall thickness in mouse heart with echocardiography, since even clinically significant myocardial edema results in a 10% increase in ventricular wall thickness, which corresponds to 1 mm in adult human heart (24). We measured the myocardial water content using wet-to-dry weight ratio in mice and observed that myocardial water content increased by 8.7% at 1 week after MI. We also evaluated the post-infarction cardiac dysfunction with echocardiography and found that left ventricular function is maximally reduced at 1 week post MI and slightly recovers 4 weeks later. Our finding coincides well with the time course of the postinfarction myocardial edema reported by Nilsson et al. (25), who measured the myocardial edema using the magnetic resonance imaging in human. They found that myocardial edema was greatest 1 week after the infraction with gradual decline during the following months, but lasted surprisingly long with a median duration of 6 months. Based on the report demonstrating that 3.5% increase in myocardial water content resulted in 40% decrease of cardiac output (26), we can assume that the degree of myocardial edema developed in our study is more severe.
Despite the presence of several AQPs in myocardium, their roles in development and resolution in myocardial ischemia have not been established yet. We demonstrated here the up-regulation of cardiac AQPs following MI. However, time-dependent patterns in their expression were different. The time course of AQP4 expression correlated well to that of left ventricular dysfunction which is maximal at 1 week followed by a partial recovery at 4 weeks, whereas AQP1 and AQP6 increased steadily with time after MI irrespective of cardiac function. Warth et al. (27) also observed upregulation of AQP4 mRNA and protein after 2 hr of ischemia and 3 hr of reperfusion, but showed anti-AQP4 immunoreactivity decorating the whole sarcolemma of ischemic myocytes. In addition to the expression level, we found MI-induced changes in the distribution pattern of AQP4 proteins. The pattern of anti-AQP4 staining at the intercalated discs has changed to give more undulating appearance following MI, presumably due to integrity loss of the intercalated discs. The dissipation of gap junction and remodeling of intercalated discs was frequently observed following MI (28). As AQP4 is a well-known player for development of cellular edema following cerebral ischemia (29), polarized localization of AQP4 is worthy of note for myocardial water and ionic homeostasis. It was suggested that AQP4 in astrocyte endfeet not only closely associates but also functionally interacts with the inward rectifying potassium channel, Kir4.1, for extracellular K+ buffering (30). In cardiac tissue, AQP4 might also work in concert with adjacent ion channels and gap junctions, and help to maintain the constant volume and ionic concentration of interstitial fluid at intercalated discs by facilitating water transport across the membrane for normal excitation and conduction or for the tolerance against ischemic insults.
MI-induced increase of AQP1 expression was not as strong as the other two AQPs, and mainly confined to the blood vessel, which was supportive of the previous findings (31). The abundance of AQP1 protein in cardiac tissue after MI reflects the increased numbers of microvasculature in response to hypoxia and might contribute to the interstitial fluid accumulation. Implication of AQP1 for the development of lung edema has been described (32). MI-induced increase of AQP6 expression was the most prominent among the AQPs tested, and gave a clear and distinct striation pattern to cardiac myocytes with the widening of sarcomeres and the space between them, presumably due to cell swelling. After MI, accumulation of acid metabolites such as lactate due to the death of ischemic myocytes would induce acidification of cytoplasm pH, which might trigger AQP6 to permeate anions including nitrate, a metabolite from nitric oxide. However, the role of AQP6 in cardiac ischemia and post-infarct edema formation, either as a water channel or an anion channel, needs to be elucidated.
The possibility cannot be excluded that the observed up-regulation of AQP1, AQP4, and AQP6 at the early stages of ischemia might act as a protective mechanism of heart from developing post-infarct edema via facilitating water shift for its drainage towards lymphatics. However, it might more reasonable to explain AQP up-regulation as a contributing factor to developing post-infarct edema as the inwardly directed osmotic gradients for water flow would be created due to the rapid accumulation of acid metabolites after MI (33). To get more insight into the role of AQPs in myocardial edema formation, further experiments will be necessary to evaluate the levels of cardiac AQPs in the diverse pathologies of the heart associated with exceeding water accumulation in the myocardium using subtype-specific AQP null mice. Manipulating the expression levels of AQPs to avoid cell swelling might be a possible strategy for the cardiac protection and ischemic preconditioning.




 XML Download
XML Download