

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Despite the diverse etiologies of renal parenchymal calcification (nephrocalcinosis), nephrocalcinosis combined with electrolyte imbalance of hypokalemia can narrow down the differential diagnosis into hereditary tubulopathy such as Bartter's syndrome, chronic laxative or diuretic abuse, type 1 renal tubular acidosis (RTA), and recently recognized acute phosphate poisoning such as oral sodium phosphate bowel preparation (OSPBP) (1, 2).
Since the first introduction of an unusual form of cortical nephrocalcinosis due to "acute phosphate nephropathy" by Desmeules et al. (2) in 2003, OSP-BP associated acute or chronic renal injury has been documented in several other reports (1, 3). Also, transient hypocalcemia and hypokalemia after OSP-BP was noted as electrolyte imbalances in more than 50% of elderly patients (4). In contrast, the prominent clinical features of type 1 RTA are characterized by medullary nephrocalcinosis/urolithiasis and bone involvement due to sustained hypokalemia and metabolic acidosis.
In the present case, the unique clinical experience of acute phosphate poisoning after OSP-BP leading to the disclosure of previously unrecognized chronic type 1 RTA due to Sjögren's syndrome is introduced.
CASE DESCRIPTION
A 30-yr-old woman presented to the emergency department (ED) in extreme mental agitation with the recent onset of carpal spasm, a tingling sensation in the lower extremities, and circumoral numbness about 2 hr after the second dose of oral sodium phosphate (OSP) on March 14, 2012, which she took for the bowel preparation of a screening colonoscopy. She had watery diarrhea twice with abdominal discomfort before presentation to ED.
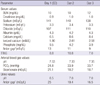
In ED, vital signs were stable but with a respiratory rate of 24/min, and other physical examinations were unremarkable except for the presenting neuromusclar manifestations of hypocalcemia including the positive Chvostek's sign. As shown in Table 1, laboratory findings with normal kidney function in ED were hyperphosphatemia (9.5 mg/dL; normal, 2.5-4.5 mg/dL), hypocalcemia (total calcium of 6.5 mg/dL and ionized calcium of 1.90 mM/L; normal, 2.26-2.64 mM/L), hypokalemia (3.3 mEq/L), and respiratory alkalosis (alkaline arterial pH, 7.53; bicarbonate, 21.1 mEq/L; PCO2, 24.8 mmHg). After the intravenous injection of 1 gram of calcium gluconate in ED, carpal spasm resolved rapidly, and hyperphosphatemic hypocalcemia was no longer noticed. On subsequent hospital days (day 2 and 3), however, sustained hypokalemia (3.3 to 3.4 mEq/L) with inappropriately high transtubular potassium gradient (≥ 7.0) suggesting renal potassium wasting despite dietary potassium supplementation, metabolic acidosis with normal blood anion gap (9 to 11 mEq/L; reference, 10 ± 2 mEq/L), and alkaline urine pH (> 6.0) with positive urine anion gap (> 16 mEq/L) suggested type 1 RTA, which was ascertained by other positive evidences for the impaired urinary acidification of distal tubules by loopdiuretic (torsemide) test (urine pH > 6.5) and urine to blood PCO2 gradient (> 20 mmHg) upon intravenous alkali load with sodium bicarbonate.
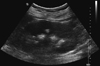
On further questioning in careful history taking, she admitted chronic gritty sensation of eyes using artificial eye drops frequently and dry mouth with polydipsia and polyuria more than 5 yr, which suggested an impaired exocrine gland secretion mostly due to Sjögren's syndrome. Also, keratoconjunctivitis sicca was confirmed by Schirmer's test. Furthermore, several autoantibodies for Sjögren's syndrome were positive with high titers; antinuclear antibodies, antibodies to the ribonucleoprotein antigen La (SS-B) and Ro (SS-A), and rheumatic factor. In imaging tests, the subsequent abdominal ultrasonography revealed prominent medullary nephrocalcinosis (Fig. 1), and the salivary scintigraphy showed the evidence of salivary gland dysfunction in both submandibular and parotid glands (Fig. 2). Therefore, the secondary type 1 RTA due to Sjögren's syndrome was diagnosed. Subsequently, oral intake of potassium citrate (Urocitra-K®, 1,080 mg/tablet) was initiated, of which dose was adjusted for the adequate alkali supplementation as well as for the correction of hypokalemia. Since then, hypokalemia and metabolic acidosis were no longer noticed.
DISCUSSION
Carpal spasm in this patient is most likely caused by hypocalcemia because of the transient hyperphosphatemia leading to the precipitation of calcium-phosphate complexes with acute phosphate poisoning after the use of OSP-BP. The supportive evidence is the presence of hyperphosphatemic hypocalcemia on presentation with the complete resolution of the symptoms and hyperphosphatemia within 2 hr of admission (Table 1). Also, the acute respiratory alkalosis (pH 7.53, PCO2 24.8 mmHg) as a result of the extreme mental agitation caused by the presenting neurological symptoms probably further contributed to a reduction in free ionized calcium concentration (5).
Because of the lower cost and the better tolerance, OSP remains popular in the bowel preparation for colonoscopy in clinical practice. However, the bowel preparation using OSP is often associated with mild to severe electrolyte imbalances, such as hyperphosphatemia, hypocalcemia, hypernatremia, hyponatremia, hypokalemia, and high anion-gap metabolic acidosis. Even acute and subsequent chronic renal injury due to "phosphate nephropathy" has been observed (1). Deposition of calcium-phosphate crystals in distal renal tubules after OSP can be only proven by renal biopsy since the intrarenal deposition in acute hyperphosphatemia is usually not sufficient to be detectable in radiological imaging (1-3, 6). Furthermore, nephrocalcinosis associated with acute phosphate nephropathy is more predominantly localized in the renal cortex than medulla. The medullary nephrocalcinosis as in this case (Fig. 1) is seen usually with sustained hypercalcemia and/or hypercalciuria related to malignancy, hyperparathyroidism, furosemide or laxative abuse, sarcoidosis, Type 1 RTA, etc (3).
Following the admission, as shown in Table 1 and Fig. 1, sustained hypokalemia with renal potassium loss (high transtubular potassium gradient (TTKG) ≥ 7.0), normal anion-gap metabolic acidosis, the persistent urine pH > 6.0 in the presence of metabolic acidosis, and medullary nephrocalcinosis suggested the impaired distal urinary acidification due to type 1 RTA (7). The classical test using oral ammonium-chloride loading to assess the impaired distal urinary acidification was not performed because of the substantial pre-existing systemic acidemia. Instead, the oral administration of torsemide rather than furosemide, recently known to be more specific in assessing urinary acidification of the distal nephron in a study, showed sustained alkaline urine pH more than 6.5 (8). Furthermore, the sodium bicarbonate (NaHCO3) loading test also revealed the urine-to-blood PCO2 gradient that was persistently less than 11.2 mmHg when the urine pH (7.62) exceeded the corresponding blood pH (7.39) (9). Hence, there is little doubt for the certainty of the diagnosis of type 1 RTA in this patient.
Approximately 10% of the patients with Sjögren's syndrome have clinically significant renal disease. The renal involvement of the disease is often associated with RTA (33%), predominantly type 1 RTA and less frequently type 2 RTA (10). In an immunocytochemical study, the absence or decrease of H+-ATPase in the intercalated cells of cortical collecting tubules of distal nephron has been shown, and appears to play an important role in the pathogenesis of type 1 RTA in patients with Sjögren's syndrome (11).
Is it likely that the underlying type 1 RTA contributed to acute hypocalcemia? Chronic metabolic acidosis results in negative calcium balance due to the renal wasting of calcium (12). Although serum calcium is usually maintained within normal limits because of the compensating secondary hyperparathyroidism, the ionized calcium concentration in the serum must be lower than usual in the presence of chronic metabolic acidosis in order to induce secondary hyperparathyroidism. Thus, any additional factor that reduces serum calcium such as acute hyperphosphatemia as in this case would have a greater hypocalcemic effect in an untreated patient with type 1 RTA.
The major underlying mechanism of nephrocalcinosis in type 1 RTA is metabolic acidosis: chronic metabolic acidosis stimulates bone resorption and impairs renal tubular reabsorption of calcium, both of which contribute to hypercalciuria; nephrocalcinosis is further aggravated by a low urinary citrate excretion, which in the case of type 1 RTA is due to metabolic acidosis and hypokalemia; metabolic acidosis leads to hypokalemia because of the increased delivery of sodium to the cortical collecting duct and the stimulation of aldosterone (5, 13, 14). Hence, the mainstay of the treatment for nephrocalcinosis is to prevent its progression by correcting the biochemical abnormalities associated with type 1 RTA.
The presence of hypokalemia in type 1 RTA by inducing low intracellular pH of the proximal tubular cells contributes to decreased excretion of urinary citrate (14). That explains why urinary citrate excretion is not reduced in type 4 RTA. In fact, the increase in citrate excretion following the administration of potassium citrate is the result of correction of metabolic acidosis, not the citrate administration per se. Although potassium bicarbonate for the therapy of type 1 RTA can be equally effective, it causes more side effects, such as gas formation of CO2 in the stomach as bicarbonate reacts with HCl. Therefore, potassium citrate by either as a drug or from natural food sources including beverages would be an ideal substance for adequate alkali supplementation (1-2 mM/kg/d in adults) and for the correction of hypokalemia and hypocitraturia as well.
In summary, the precipitation of calcium-phosphate complexes by acute hyperphosphatemia after OSP-BP caused acute hypocalcemia, which was further aggravated by acute respiratory alkalosis, leading to the manifestation of hypocalcemic tetany. Subsequently, the sustained hypokalemia, nephrocalcinosis in medulla, and biochemical findings of impaired ability of urinary acidification unveiled the secondary type 1 RTA due to Sjögren's syndrome.

 XML Download
XML Download