

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Familial Mediterranean fever (FMF) is an autosomal recessive disease characterized by recurrent episodes of fever accompanied by peritonitis, pleuritis, arthritis, or erysipelas-like erythema (1). It is known to occur mainly among Mediterranean and Middle Eastern populations such as non-Ashkenazi Jews, Arabs, Turks, and Armenians. FMF is not familiar to clinicians beyond this area and diagnosing FMF can be challenging. We report a case of FMF associated with multiple venous thrombosis and elevated immunoglobulin D (IgD) level diagnosed by MEFV mutation analysis.
CASE DESCRIPTION
A 22-yr old man was referred to our hospital on February 17, 2012 due to fever and arthalgia which started 2 days prior to admission. He had a history of recurrent episodes of fever associated with arthalgia lasting 5 days with 5-6 month intervals which started at the age of 2. Fever would subside spontaneously or with the use of antipyretics.
On admission he showed a fever of 40℃ with abdominal pain and arthalgia in both knees. Laboratory findings on admission showed mild leukopenia and thrombocytopenia (white blood cell count of 2,650/µL and platelet count of 102,000/µL). Erythrocyte sedimentation rate was normal but C-reactive protein was highly elevated at 27.88 mg/dL.
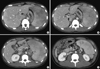
Chest and abdomen computed tomography (CT) was performed as part of the diagnostic work up for fever of unknown origin. Chest CT showed no specific findings while abdomen CT showed thrombosis in portal vein (PV) and superior mesenteric vein (SMV) with splenomegaly. On the 10th hospital day, the patient complained of left upper quadrant pain and 3D angio CT showed total thrombosis of splenic vein with partial thrombosis of proximal SMV, main PV and intrahepatic both PV with small amount of ascites (Fig. 1). Bone marrow biopsy and lumbar puncture results were normal.
Considering the patient's history of periodic fever with no familial history, autosomal recessive periodic fever syndromes such as FMF and Hyperimmunoglobulin-D syndrome (HIDS) were suspected. IgG, IgA, and IgM were all in the normal range while IgD was elevated at 39.2 mg/dL. The patient also satisfied the three major criteria (fever, abdominal pain, joint pain) of the Tel-Hashomer criteria. To verify the diagnosis, DNA analysis of the MVK and MEFV gene was performed. MVK for HIDS was negative while 3 mutations (p.Glu148Gln, p.Pro369Ser, p.Arg408Gln) were identified in the MEFV gene for FMF. After diagnosis of FMF, colchine was prescribed and the patient is currently without recurrent fever.
DISCUSSION
In recent years, more and more cases of FMF have been reported outside the Mediterranean or the Middle Eastern countries, such as the US and Japan. In Japan, more than 90 cases of FMF were reported (2, 3). In Korea, however, only one case of FMF has been reported, in a 8 yr-old boy associated with renal amyloidosis (4).
The 4 main mutations found in the studies that identified the MEFV gene responsible for FMF were p.M680I, p.M694V, p.M694I, and pV726A on exon 10 (5, 6). Subsequently, the p.E148Q sequence alteration was identified on exon 2 (7). Several studies have pointed out that these 5 mutations are responsible for more than 85% of FMF patients in the Middle Eastern Area (1).
Our patient showed complex mutations (p.Glu148Gln, p. Pro369Ser, p.Arg408Gln) in the MEFV gene. A significant number of patients diagnosed as FMF have only a single mutation despite sequencing of the entire MEFV genome region or other autoinflammatory genes, and this has led to a reconsideration of the simple loss of function of the recessive model of FMF inheritance (8, 9). In some cases, FMF can be viewed as a dominant condition with low penetrance and variable disease expression, presenting not only in homozygous subjects, but also in heterozygous subjects (10). Over 200 sequence variants have been recorded in the database of the Registry of Hereditary Auto-Inflammatory Disorders Mutations (http://fmf.igh.cnrs.fr/infevers) and the same complex mutation as our patient could be found in an Armenian patient. It remains controversial whether the E148Q mutation is a disease-causing mutation or a simple polymorphism because of high allele frequency in healthy controls (11). Topaloglu et al. (12) reported that both homozygous and compound heterozygous patients for E148Q were symptomatic and E148Q mutation was described as producing a milder FMF phenotype with low penetrance (13). Therefore, it is likely that this mutation can be a cause FMF. MEFV mutation analysis on Korean patients is yet to be studied, as our case is the first adult case to be reported in Korea.
HIDS was considered as the diagnosis due to the high level of serum IgD. However elevation in serum IgD has been described in other autoinflammatory diseases (14) and although different from our mutation, Medlej-Hashim et al. (15) reported elevated IgD plasma levels associated with homozygotic status for M694V, and to a lesser extent V726A.
Hypercoagulability has been recently described in patients with FMF, with major thrombotic events occurring with amyloidosis (16). FMF presenting with pulmonary embolism (17) and FMF complicated by Budd-chiari syndrome (18) have been reported but this is the first case presenting with thrombosis in PV, SMV and splenic vein. It has been reported that decreased protein S activity is related to thrombotic disorder in systemic inflammatory disease such as Behcet's disease patients (19) and protein S activity was decreased in our patient. There are no reports of HIDS associated with thrombosis.
In conclusion, we report a 22-yr old male diagnosed with FMF despite high level of serum IgD. The diagnosis of FMF does not require a MEFV mutation analysis but clinical diagnosis of FMF is not easy and analysis of MEFV mutation can be helpful, especially in patients of non-Mediterranean origin. When thrombosis is associated with elevated IgD, FMF should be suspected. This is the first adult case of FMF in Korea.
 XML Download
XML Download