

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Medullary thyroid carcinoma (MTC) is an uncommon malignant tumor derived from calcitonin-producing C cells of the thyroid. Seventy five percent of all MTCs are sporadic, while the remaining 25% of MTCs occur in three well-defined familial syndromes; multiple endocrine neoplasia (MEN) 2A, MEN 2B, and familial medullary thyroid carcinoma (FMTC) (1). The inherited forms of MTC are autosomal dominant traits associated with point mutations of the RET proto-oncogene, which encodes a receptor tyrosine kinase that plays roles in the regulation of cell proliferation, migration, and differentiation (2).
In the literature, RET proto-oncogene mutations of MEN 2A and FMTC in Koreans have been observed most commonly in codons 634 and 618, and rarely in codons 630, 631, and 768 (3-5). However, MTC with codon V804M RET mutations has not reported in Korea. In the present study, we screened a Korean family in which one member had a rare V804M RET germline mutation that was detected by genetic analysis.
CASE DESCRIPTION
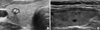
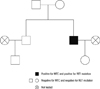
A 33-yr-old man (index patient) was incidentally diagnosed with a 0.7 cm sized nodule in the right thyroid at a general check-up on 30 January 2012 (Fig. 1). He had no personal or family history of hypertension or thyroid cancer. Physical exam was normal. Ultrasound (US) guided fine needle aspiration (FNA) was performed and was suspicious for MTC. Laboratory testing revealed serum calcitonin 23.9 pg/mL (normal range < 10 pg/mL), CEA 5.77 ng/mL (normal range < 5 ng/mL) Ca 9.4 mg/dL, P 3.9 mg/dL, and intact parathyroid hormone (iPTH) 27 pg/mL (normal range 15.0-65.0 ng/mL). The patient underwent total thyroidectomy with central compartment neck dissection for the thyroid tumor. Histological examination revealed MTC without metastasis. Genomic DNA was extracted from the peripheral blood leukocytes of both index patient and at-risk family members. Full sequencing of the RET gene and analyses of exons 10, 11, 13, 14, 15, and 16 were subsequently performed. Genetic analysis of the index case revealed a missense GTG → ATG mutation at codon 804 in exon 14 of the RET proto-oncogene by single-strand conformation polymorphism analysis and direct sequencing using an automated DNA sequencer (3130 Genetic Analyzer, Applied Biosystems, Foster City, CA, USA). This transversion leads to the substitution of valine with methionine (Fig. 2). At-risk family members were tested for exon 14. No other family members carried the V804M RET mutation (Fig. 3). Biochemical tests and thyroid US examinations were performed on family members. On thyroid US, the index patient's mother had multiple small nodules on her left thyroid gland (Fig. 1), but US-guided FNA cytology revealed benign. Laboratory findings including serum Ca, P, iPTH, and calcitonin were within normal limits in all family members.
Prior to obtaining samples, all individuals included in the study were fully informed about the genetic study and consent was obtained.
DISCUSSION
Clear associations between RET mutation genotype and phenotype have been reported in hereditary MTCs. MEN 2A is caused by mutations affecting cysteine residues in codons 609, 611, 618, and 620 within exon 10 and codon 634 in exon 11 of RET (6). In MEN 2B, the mutation in codon 918 in exon 16 is present in over 95% of patients, while codon 883 in exon 15 of RET is present in 2%-3% of patients (6). Rarely, patients with MEN 2B have double RET mutations including V804M and V805K, V804M, and Y806C (7, 8). FMTC is most commonly associated with mutations in codons 609, 611, 618, and 620 in exon 10, codon 768 in exon 13, and codon 804 in exon 14 (6).
Thus far, mutations at the 804 codon have been reported in less than 2% of cases (6, 9). Two types of codon 804 RET mutations have been reported, V804L and V804M. Phenotypes associated with the V804M RET mutation have been reported almost exclusively in FMTC. Only several cases of V804M mutations associated with MEN 2A (MTC plus hyperparathyroidism) have been reported in the literature (10-12).
FMTC is now viewed as a phenotypic variant of MEN 2A, with decreased penetrance for pheochromocytoma and primary hyperparathyroidism, rather than as a distinct entity (13). The most rigid definition of FMTC is multigenerational transmission of MTC in which no family member has either pheochromocytoma or primary hyperparathyroidism (14). A less rigid definition is the presence of MTC in four affected family members without other manifestations of MEN 2A (7). Therefore, a family with a small number of MTC-affected individuals who appear to have FMTC could mask the eventual identification of pheochromocytoma (15). We initially suspected our case to be FMTC because he had MTC with a germline mutation but without manifestations of MEN 2A. However, there were no germline mutations in other members of his family. Therefore, our case was a de novo V804M RET mutation carrier.
The American Thyroid Association (ATA) guidelines stratify all known RET mutations into one of four risk levels (ATA risk levels A-D) (13). ATA level D mutations carry the highest risk for MTC and ATA level A mutations carry the "least high" risk. ATA-A mutations include RET gene mutations at codons 768, 790, 791, 804, and 891 (13). Despite the ATA categorization into four levels (A-D), differences in the development and behavior of MTC and the development of MEN 2A features are present between RET mutations even within the same ATA level (16). Some researchers have reported that V804L or V804M mutations have more aggressive potential (17, 18). Our patient was already 33 yr of age and had developed microcarcinoma without lymph node metastasis; therefore, this case does not appear to have aggressive potential.
ATA recommends that all patients with personal medical histories of primary C cell hyperplasia, MTC, or MEN 2 should be offered germline RET testing (13). Once a germline RET mutation has been identified in a family, RET mutation analysis should be offered to all first-degree relatives of known mutation carriers before the age of recommended prophylactic thyroidectomy whenever possible (13). Clinical implications, ATA risk level and prophylactic thyroidectomy testing and therapy according to the genotype of RET mutations are summarized in Table 1.
Most laboratories evaluate patients for mutations in the five most commonly mutated codons in exons 10 and 11 (C634R, C609, C611, C618, and C620) (6) and additionally sequence exons 13, 14, 15, and/or 16. Some laboratories use a two-tiered approach to the analysis, starting with sequence analysis of the most commonly mutated "hotspot" exons and, at the request of the ordering physician, sequence the remaining exons of RET if the initial analysis is negative (19). Tiered approaches risk failure to detect rare double mutations. For example, there are a few reports suggesting that codon 804 mutations in conjunction with a second variant in RET are associated with FMTC (20) and MEN 2B (7, 8). We performed a full sequencing analysis of RET proto-oncogene mutations for exon 10, 11, 13, 14, 15, and 16 on the index patient. No double RET mutation was noticed in index patient.
The prevalence of V804M is very high among the residents of Sardinia, an Italian island. Pinna et al. (10) reported the prevalence of V804M in up to 48% of index cases (3/7 families) and 59% of overall family members (17/19 relatives) in Sardinia, possibly as a consequence of founder effect in this population. Shifrin et al. (12) also reported 107 members of an Italian family (origin from Florence/Calabria) with RET V804M proto-oncogene mutations. With exception of Italian, RET V804M protooncogene mutations are very rare in other races. In Asia, only one case of V804M and Y806C in the same allele in patient with MEN 2B was reported (8). However, V804M RET mutations have not been previously reported in Korea. Our case is the first in Korea, and represents a de novo V804M RET mutation.


 XML Download
XML Download