

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The Long QT syndrome (LQTS) is a rare hereditary disorder characterized by prolonged QT interval on electrocardiogram (ECG). Affected individuals have risks of fatal ventricular tachycardia (VT), torsades de pointes (TdP) and sudden cardiac death (SCD) (1, 2). The cluster of disease was first recognized in a family in which several children with congenital hearing loss revealed QT prolongation on ECG, and have experienced recurrent syncope and SCD, with an inheritance of autosomal recessive manner (Jervelle and Lange-Nielsen [JLN] syndrome) (3). Following the report, similar disorder with familial cluster that showed QT prolongation and recurrent syncope, but without deafness was reported independently in two different families with an autosomal dominant inheritance (Romano Ward syndrome) (4, 5).
This disease is now understood as a cardiac channelopathy in which ventricular repolarization is delayed due to the prolonged duration of myocardial action potential. In LQTS, the functions of several ion channels or related intracellular structures are impaired and the net outward flow during cellular repolarization is decreased. This decreased outward ionic current results in QT interval prolongation on ECG, and this is associated with early afterdepolarization that may potentiate triggered activity to initiate a VT. Sodium (Na), potassium (K) and calcium (Ca) are the three most important ions that determine the duration of cellular action potential. To date, mutations in 12 different genes that encode channel subunits or intracellular structures associated with these ionic current have been found (6).
To quantify the clinical features of disease, a weighted scoring system has been proposed to use in clinical settings. Long QT (LQT) score or Schwartz score was first developed in 1985 and was revised in 1993 (7, 8). The scoring system includes various clinical features such as ECG findings (QTc, T wave morphology, presence of bradycardia, and TdP), clinical findings (syncope with or without stress) and family history (sudden cardiac death at young age, LQTS). And the probability of LQTS is defined as low, intermediate and high for the score of < 1, 2 to 3 and ≥ 4, respectively.
The International Long QT Syndrome Registry was founded in 1979, and various clinical and genetic studies, including many prospective studies, have been extracted from this registry (9, 10). In Korea, however, there have been only a few studies for LQTS regarding clinical and ECG findings (11) or genomic sequences (12) only in small study populations. In this study, we aimed to clarify the clinical and genetic characteristics of Korean LQTS population including children and adolescents from a single institution at the point of February 2012.
MATERIALS AND METHODS
We searched for the patients who had the diagnosis of LQTS on electronic medical record (EMR), or who had undergone genetic testing for long QT gene in Seoul National University Hospital (SNUH) at the point of February, 2012. The genetic testing to identify specific mutation for LQTS is commercially available now, and genetic studies for 3 major long QT genes (KCNQ1, KCNH2, and SCN5A) were performed sequentially to detect affected population in SNUH. Genetic testing for other relatively infrequent mutations such as KCNJ2 (LQT7) or CACNA1C (LQT8) was performed for specific requests.
Proband group
We enrolled patients with LQTS as proband group if at least one of their serial ECG showed prolonged QTc longer than 0.47 sec; or positive in genetic test for LQTS. Excluded were subjects without mutation for LQTS and any QTc longer than 0.47 sec on routine ECG, 24 hr Holter monitoring, epinephrine challenge test (drug test), or treadmill test (TMT).
The collected data include gender, age at initial symptom or presentation, age at diagnosis, current age, mode of presentation, follow-up duration, presence of symptom prior to treatment, mode of treatment, change of symptom after treatment, results of genetic test, and ECG data (on routine ECG, 24 hr Holter monitoring, epinephrine challenge test, or TMT). Schwartz score (Long QT score) was calculated with collected data for each patient.
Family group
In a part of proband group, after the diagnosis for LQTS was confirmed with genetic studies, additional genetic tests for family members were performed. Of these family members, those of whom genetic tests revealed positive results of mutation for long QT gene were included in the family group.
We calculated QTc at lead II, V2, and V3 of serial ECGs with Bazett formula, and the longest QTc value was adapted for analysis.
Statistical analysis
Statistical analysis was performed using the Student's t-test for continuous variables and the chi-square test for nominal variables (Statistical Package for the Social Sciences [SPSS], version 20.0 [SPSS Inc., Chicago, IL, USA]). A P value of < 0.05 was considered statistically significant.
RESULTS
Proband group
We identified a total of 71 patients who had the diagnosis of possible LQTS on EMR. Among those, one patient experienced ventricular tachycardia with prolonged QTc on ECG (0.491 sec), but she was not selected for proband group as she had intracardiac mass around inflow area of right ventricle. Other 8 candidates were excluded because of insufficient evidences of LQTS. Finally, a total of 62 subjects was included in proband group. Fifty one patients were followed up at the Department of Pediatrics, and 11 at the Department of Internal Medicine.
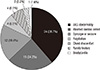
In proband group, the median age was 15.8 yr (1.4-86.2 yr). The age distribution at initial symptom or diagnosis in this group is shown in Fig. 1. Thirty four were female and 28 were male. There was no statistically significant difference in sex distribution (P = 0.451). In symptomatic subjects, excluding the subjects with incidental ECG findings from the proband group, the median age at initial symptom was 10.5 yr (0.0-70.3 yr). The modes of presentation were aborted cardiac arrest (n = 15, 24.2%), syncope or seizure (n = 12, 19.4%), palpitation (n = 4, 6.4%), chest discomfort (n = 4, 6.4%), family history of probable LQTS (n = 2, 3.2%), bradycardia (n = 1, 1.6%), and incidentally found ECG abnormality (n = 24, 38.7%) (Fig. 2). There was only a pair of siblings in a family, and all the other subjects in proband group were unrelated.
The mean value of longest QTc was 0.539 ± 0.064 sec (0.419-0.784 sec). The longest QTc was longer than 0.470 sec as our inclusion criteria in all subjects except one patient. The patient had typical clinical features of Anderson-Tawil syndrome (LQT7) and was confirmed by genetic test and prominent U wave (will be discussed later). Thirteen patients (21.0%) had notched T wave on more than 3 precordial leads. Four (6.5%) showed T wave alternans on the baseline ECG, drug test or TMT. Fourteen patients (22.6%) had the documented VT; and 10 (16.1%) experienced polymorphnic VT with documented ECG recordings. Sixteen (25.8%) were identified to have bradycardia on serial ECGs.
Total number of patients, who had ever had the experience of any syncopal attack during their lives, was twenty six (41.9% of 62). Syncopal episodes were found during exercise in 12, with emotional stress 4, during sleep 4, and under unidentified situation in 6. Fifteen patients (24.2%) experienced any aborted cardiac arrests, which occurred during exercise in 8, emotional stress in 2, sleep in 2, and under unidentified situation in 3. All these 15 patients initially presented to hospital with cardiac arrest. Three (4.8%) accompanied congenital deafness that they could be classified as JLN syndrome. Long QT score or SS was calculated in every subject of proband group. The mean value of Schwartz score was 4.4 ± 1.6 (0.0-8.5). There was only one (1.7%) patient that had the Schwartz score lower than 2. Twenty five (41.7%) patients were those with the Schwartz score of 2-3, and 23 (38.3%) with 3-4. There were 11 (18.3%) patients that had the Schwartz score of 6 or more.
There was a female patient who showed recurrent sustained VT, TdP and QT prolongation from birth. Because a tachyarrhythmia had been detected on prenatal echocardiogram during her fetal life, amiodarone was administrated to her mother during pregnancy. Repeated DC cardioversions were delivered to cease VT, and her QTc was 0.517 sec. The genetic study revealed no mutation for LQT 1 and the QTc remained prolonged during out-patient follow-up. The patient was on the management with a beta-blocker and no more symptoms developed.
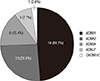
Genetic studies were performed in 42 patients (67.7%). In thirteen patients, genetic testings were done with all the 3 most important genes for LQTS (LQT1, LQT2, and LQT3 genes). Twelve patients were studied for LQT1 and LQT2 genes, and 12 for only LQT1 gene. One patient was done with LQT1 and LQT3 gene studies sequentially, and 3 patients were studied for the LQT7 gene with their characteristic symptoms. There was one patient that had undergone genetic study for LQT8. Of those, 21 (50.0% of 42) were confirmed to have the mutation of LQT gene, and 9 had the mutation in KCNQ1 (LQT1), 5 in KCNH2 (LQT2), 3 in SCN5A (LQT3), 3 in KCNJ7 (LQT7), and 1 in CACNA1C (LQT8).
Three patients had KCNJ7 gene mutation with typical clinical feature of Anderson-Tawil syndrome that showed episodes of periodic paralysis. Two were siblings and one was unrelated; and the genetic studies for the three patients focused on KCNJ7 gene revealed 2 different mutations with 1 novel mutation. Two patients with KCNJ7 gene mutation had episodic nonsustained ventricular tachycardia on Holter monitoring.
One patient with CACNA1C mutation presented with cardiac arrest during general anesthesia at the age of 2.5 yr. The genetic studies for LQT1, LQT2, and LQT3 were performed; however, no mutation was found. With his typical clinical morphology such as syndactyly, flattened nose and small teeth, we did the genetic study for CACNA1C and a mutation was found that he was diagnosed as Timothy syndrome (LQT8). A beta-blocker was prescribed for him and he has experienced no further significant symptom during the follow-up of 7.5 yr.
Three patients with congenital deafness (JLN syndrome) were all tested for the mutation of KCNQ1 and KCNH2 gene, and only 1 patient was confirmed to have mutation (compound heterozygote mutation of KCNQ1).
In proband group, 3 patients were classified as LQTS secondary to other causative conditions. Of these secondary LQTS patients, 2 were followed up for chronic renal failure and had uremic hypertrophic cardiomyopathy. Their longest QTc were 0.548 and 0.531 sec, and their Schwartz scores were 6 and 3. One of them had an episode of syncope and TdP. No genetic study for LQT gene was performed with these two patients and they did not take beta-blockers. Another one was a 15.5 yr old-male patient who presented with cardiac arrest after intravenous meperidine (Demerol®) administration. Although the QTc on routine ECG was normal, epinephrine challenge test revealed QT prolongation (QTc 0.513 sec) without further development of VT or TdP. Genetic studies for LQT 1, 2, and 3 genes were performed but single rare polymorphisms were found in LQT 2,3 genes (13).
The mean duration of follow-up in proband group was 4.2 ± 3.5 yr (0.0-16.8 yr). Some without overt symptom were lost during follow-up. Beta-blockers were the most frequently applied treatment, 41 out of 62 (66.1%) were prescribed for beta-blockers. All genetically confirmed patients were on the management of beta-blockers except two patients of Anderson-Tawil syndrome. One with the mutation of SCN5A gene initially took a beta-blocker, but because another episode of cardiac arrest had developed, this LQTS patient got implantable cardioverter-defibrillator (ICD) implantation. For recurrent ICD shocks were delivered in spite of the management with beta-blocker, mexiletine was added on her medication. A total of 6 patients was implanted with ICDs, 4 were children including 3 with SCN5A mutation, and ICDs were implanted epicardially in 2 patients (Table 1). Of these 6 patients with ICDs, 2 adult patients were implanted with ICDs just after first episode of cardiac arrest which was the initial presentation of their disease, and one of them experienced ICD shocks with poor compliance for beta-blocker. In contrast, the remaining 4 pediatric patients experienced major cardiac events or ICD shocks repeatedly even during the period of the medication with beta-blockers.
Family group
Nineteen family members of 12 probands were genetically identified for LQTS. ECGs were available in 16 family members, and QTc was prolonged than 0.470 sec in 9 (56.3% of 16). Notched T wave was found in 3, and other 3 family members showed bradycardia on ECGs. Three family members (15.8%) had experienced any episode of syncope, and two of them had the mutation in KCNQ1 and one in KCNH2. All these three family members were parents of the proband, and they started the therapy with beta-blocker without any further provocation test after genetic confirmation. None of the family group had the history of ventricular tachycardia or TdP with ECG documentation.
The mean Schwartz score was 3.3 ± 1.7 (1.0-6.0). Defining LQTS as 4 or more of Schwartz score, 11 (57.9%) could be diagnosed as having LQTS. Beta-blockers were prescribed for 6 subjects in family group (31.6%) including 3 overtly symptomatic ones. None of them had any episode of cardiac arrest or was operated on ICD implantation.
Risk factors affecting the development of major events
We investigated the association of various factors with major cardiac events such as syncope, ventricular tachycardia and aborted cardiac arrest. First of all, a few ECG data were analyzed. The QTc was longer in those who had experienced syncope, VT or cardiac arrest with statistical significance (Table 2). The QTc longer than 0.508 sec could be a cut-off value for major cardiac events (sensitivity 0.806, specificity 0.600). The presences of notched T wave, T wave alternans or bradycardia were not associated with the development of syncope (P = 0.126, P = 0.276, P = 0.559, respectively) or VT (P = 0.128, P = 0.319, P = 0.123, respectively). However, the patients with the history of cardiac arrest would have T wave alternans (P = 0.013) or bradycardia (P = 0.011).
Gender, the age of initial presentation or the presence of genetic mutation had no association with the development of major cardiac symptoms (P = 0.317, P = 0.942, P = 0.180, respectively). Schwartz score could be a predictor for the development of VT or cardiac arrest. Subjects with 4.0 or more Schwartz score would have experienced VT or cardiac arrest than those with Schwartz score < 4.0 (Table 3).
As shown above, there was no gender difference in proband group and this was also true when LQTS patients were defined as having Schwartz score of 2 or more (P = 0.260), or 4 or more (P = 1.000). There was no statistical difference in QTc, Schwartz score, the age of initial presentation, cardiac events between genders. Dividing the subjects into two group for the age of initial symptom development by the age of 13 yr, male was predominant in group with less than 13 yr (M:F = 15:9) and female in that with 13 or more (M:F = 2:9), and this was statistically meaningful (P = 0.014).
Of total 81 probands and family members, 40 (49.4%) were identified to have the mutation of LQT gene. When LQTS was defined as having 4 or more Schwartz score, overall penetrance of genetic mutation was 70.0% (28 out of 40) in whole study population. The penetrance was 57.9% (11 out of 19), when calculated separately in family group with the same definition.
Distributions of involved genes are illustrated on Fig. 3. The presence of mutation was associated with the overt LQTS (Schwartz score ≥ 4.0) but this association was no longer present after separating the population who have not undergone genetic study (P = 0.096). The mutation had no association with QTc and presences of major cardiac events. The age of initial symptom was younger in the group with proven mutation (Table 4).
Beta-blockers were taken by 47 out of total 81 subjects (58.0%). Of 36 symptomatic subjects, 29 (80.6%) were on management with beta-blockers, whereas 18 of 45 (40.0%) took beta-blockers in asymptomatic group. Of 29 symptomatic patient with beta-blockers, only 5 patients experienced additional symptoms after the initiation of medication. All of these were operated on ICD implantation. Beta-blockers were not completely effective on the symptomatic patients with SCN5A mutation that all of them experienced cardiac event or ICD shocks during medication and some needed additional mexiletine. Of the 6 patients who were refractory to beta-blockers, 3 were confirmed as having SCN5A mutation and the other 3 had no information for that mutation. The longest value of QTc on the sequential ECG was significantly reduced after the initiation of medication (0.5361 ± 0.0792 sec vs 0.4916 ± 0.0610 sec, P = 0.007).
DISCUSSION
The LQTS is a potentially lethal disease, with which the patients manifest various presentations and symptoms. This is a genetically heterogeneous group with disorders in cardiac repolarization. There have been extensive clinical and genetic studies from the International Long QT Syndrome Registry, but only a few studies were performed on Korean population. This is the first study to investigate the clinical and genetic characteristic of LQTS in Korea.
In our study, the median age of first episode of symptom was 10.5 yr. There have been other studies that described the mean age of initial symptom as childhood or early adulthood (14, 15). In our study, 2 patients presented first episode before the age of 1 month, and 3 did before 1 yr.
For LQTS is a disease inherited with autosomal dominant or recessive trait, and there is no gender difference in patient population. In our study, there was no gender difference of population, either. There was no significant difference in various clinical features or ECG findings between two genders. However, when divided into two groups by the age of 13 yr of initial symptom, male was predominant in younger group and female in older group. Similar results have been observed in other studies (16, 17) and this risk associated with gender difference is thought to have some relation to hormonal factors, especially sex hormones such as androgen and estrogen (9, 18, 19).
The QTc on routine ECG, drug test or TMT was identified to be a good predictor for the development of symptoms such as syncope, VT or cardiac arrest in our study. There have been consistent data which show baseline QTc interval ≥ 0.500 sec is associated with a high risk of cardiac events (9, 16, 20, 21). In our study, the QTc longer than 0.508 sec may be used as a cut-off value for major cardiac events (sensitivity 0.806, specificity 0.600). We observed that QTc intervals on serial routine ECGs had dynamic values even in same patient. Some patients did not reveal QT prolongation on the routine ECG, but uncovered the hidden QT prolongation only with drug test or TMT. We must recognize that quite a lot portion (21.7%) of symptomatic patient presented their initial symptom as a cardiac arrest. Because missing the correct diagnosis for LQTS could result in a tragic outcome, repetitive ECGs or further evaluations for suspicious individuals are warranted.
The genetic mutation associated with LQTS had been extensively investigated. Now, at least 12 different genes are known to be related to this disease (6). There have been many researches that described the associations between the morphology of the ST-T complex and the genotype (22, 23). And risk stratification was performed for the specific genotype in many studies, but they revealed no significant contribution of genotype to outcome (17, 24). In our study, 50.0% of patients who underwent genetic studies revealed genetic mutation. This ratio of positive result is a little lower than other studies, and it may be because parts of subjects were done just for KCNQ1, KCNH2 genes. It has been shown that the mutations of 3 major genes (KCNQ1, KCNH2, and SCN5A) occupy more than 90% of whole congenital LQTS in some studies (25, 26). In our study, the distribution was 47.5%, 25.0%, 17.5%, 7.5%, and 2.5% for KCNQ1, KCNH2, SCN5A, KCNJ7, and CACNA1C, respectively. As in other studies, the presence of genetic mutation had no association with any clinical outcome in our study. The type of involved gene had no association with the age of initial symptom or diagnosis, presences of major cardiac event, or QTc interval. It means that clinical features and risk stratification with Schwartz score can be more important than genetic study for the diagnosis and for the prediction of the outcome of individual patient. Genetic studies could be a confirmative test which will not exclude the diagnosis with negative result in probable LQTS patient, or could give additional information for the management of certain type of LQTS like LQT3.
In our study, the presence of the mutation of LQT gene seemed to be associated with the age of initial presentation without statistical significance. However, there might be a bias, because no genetic study was performed for the patients who had been followed up at the Department of Internal Medicine. Excluding these patients of Internal Medicine, the association between the presence of genetic mutation and age at initial presentation became weaker (P = 0.243).
Beta-blocker plays a key role in the management of LQTS. And now it is well known that beta-blocker reduces the development of syncope and sudden cardiac death, especially in LQT 1 (21, 27-29). In our study, beta-blockers seemed to reduce the occurrence of cardiac events in that only 5 patients on the medication experienced additional events, but the exact effect could not be statistically analyzed. We should notice that all genetically confirmed patients with ICD implantation were those with SCN5A mutation in spite of relatively low proportion of this mutation. As mentioned above, beta-blockers are known to reduce the development of major cardiac events in LQT1 and LQT2 (27-29), but there are limitations in the management of LQT3 patients because of the different mechanism for repolarization abnormality in LQT3 (30). So it seems that these patients needed additional management to protect further major cardiac events other than beta-blockers.
At the point of whether beta-blockers shorten the QTc interval, there have been a little conflicting data (30-32). Our study showed that beta-blockers significantly reduced the QTc interval on the routine ECGs, which may be applied as a measure of monitoring for the compliance of medication in LQTS patient. However, we should notice that the QTc was still prolonged (0.4916 ± 0.0610 sec) even after medication in our study. More data collection and investigation should be performed.
There are some limitations in our study. The most important limitation to this study is the small sample size. Because LQTS is a group of heterogeneous clinical characteristics and genetic disorder, the sample size was not enough to compare with each clinical or genetic subgroup. And some of the subjects have been lost during follow-up at outpatient clinic partially due to the low penetrance or because no overt symptom had developed. This resulted in making the sample size for analysis smaller. Second, as our study has a retrospective design, there were some cases that the items for data analysis were omitted on EMR. These include detailed history, family history and compliance for medication, and these made our studies incomplete.
We investigated LQTS for the clinical features and genetic characteristic in a Korean tertiary medical center. The Korean LQTS patients showed similar genetic pattern, LQT1 and LQT2 covered 75% of them. The LQTS is a potentially fatal disease which manifests a tragic cardiac arrest as the first symptom. Genetic studies can make the confirmative diagnosis and advise the management options. Weighted scoring system with clinical features could predict the development of major cardiac events. More data collection and investigation should be performed to reveal this secretive and potentially lethal disease.


 XML Download
XML Download