

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The inflammasomes are a group of cytosolic protein complexes that function as a molecular platform for caspase-1-dependent proteolytic maturation and secretion of interleukin (IL)-1β and IL-18 (1-4). Of the members of the nucleotide binding oligomerization domain (NOD)-like receptor (NLR) inflammasomes, the nucleotide-binding domain and leucine-rich repeat protein 3 (NLRP3) inflammasome is the most extensively studied. Activation of the NLRP3 inflammasome in response to diverse pathogenic and endogenous stimuli leads to the assembly of protein components of the complex: the cytoplasmic innate receptor NLRP3, the adaptor apoptosis-associated speck-like protein containing a CARD (ASC), and the effector caspase-1 (1, 4, 5). The NLRP3 inflammasome is activated by two steps: a priming step is required to induce expression of both NLRP3 and pro-IL-1β; and a second step triggers assembly of the NLRP3 inflammasome after exposure to microbial toxins and ionophores or endogenous danger molecules (6, 7). In addition, the NLRP3 inflammasome is activated during infection of macrophages with various bacterial, viral, and fungal pathogens and is required for host immune defense to these pathogenic infections (6-8). The potent inflammatory activities of IL-1β and IL-18 by inflammasome activation are pathogenic in NLRP3-associated autoinflammatory disorders (5, 9). Therefore, NLRP3 inflammasome activation is tightly controlled at multiple levels. NLRP3 inflammasome activation can lead to excessive inflammatory responses and cell death, which is potentially hazardous to host defense during a variety of bacterial infections, while numerous bacteria and their components can activate or modulate NLRP3 inflammasome activation. In this review, we will examine how the NLRP3 inflammasome is activated and contributes to host defense during diverse bacterial infections. We will further discuss current knowledge of the strategies by which various pathogenic bacterial virulence factors modulate the NLRP3 inflammasome.
THE MECHANISM OF NLRP3 INFLAMMASOME ACTIVATION
The innate immune system rapidly recognizes and provides an immediate host defense response to microbial and endogenous stimuli. The germ-line encoded pattern recognition receptors (PRRs) play an important role in recognition of pathogen- and danger-associated molecular patterns. Specific subsets of PRRs, NOD-like receptor (NLR) and the PYHIN protein families, detect molecular patterns in the cytosol and activate the formation of supramolecular complexes, termed inflammasomes. The NLRP3 inflammasome, the best-characterized family member of inflammasomes, is a multi-protein complex composed of NLRP3, ASC, and caspase-1 (1-4).
The protein structure of NLRP3 contains an N-terminal pyrin domain (PYD), a central NACHT (NAIP, CIITA, HET-E, and TP1), and a C-terminal leucine-rich region (4). A recent study with high resolution structure analysis showed that NLRP3 has a specific disulfide bond between Cys-8 and Cys-108 that may participate in regulation of NLRP3 activity by reactive oxygen species (10). After sensing signals, the three components-NLRP3, ASC, and pro-caspase-1-assemble to form a typical multimeric inflammasome complex, which results in activation of caspase-1, leading to processing of pro-IL-1β and pro-IL-18 to their mature, biologically active, forms (3-5). During this activation, the adaptor protein ASC, which contains an N-terminal PYD and a C-terminal caspase activation and recruitment domain (CARD), plays an important role in assembly of the NLRP3 inflammasome through its interaction with NLRP3 via a homotypic PYD interaction. Recent studies have reported that the ASC PYD domain is also required for its self-association, as well as interaction with NLRP3 (11).
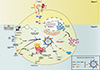
A two-signal model has been proposed for activation of the NLRP3 inflammasome. The first signal mediates transcriptional activation of pro-IL-1β and NLRP3 through recognition of ligands for toll-like receptors (TLRs), IL-1 receptor, and tumor necrosis factor (TNF) receptor. The second signal leads to inflammasome oligomerization and activation, thereby causing maturation and secretion of IL-1β and IL-18. The second signal may include a variety of pathogen-associated molecular pattern molecules (PAMPs) or damage-associated molecular patterns (DAMPs) (2, 6, 9, 12). Several important second signals have been reported; i.e., plasma membrane disruption for bacterial toxins and ATP; internalization of particulate activators by phagocytosis (1); K+ efflux and pore formation by extracellular ATP or bacterial toxins (13); or lysosomal destabilization with release of protease cathepsin B into the cytoplasm by particulate activators including silica, alum, fibrillar amyloid-β protein, or uric acid crystals (1). A recent study has shown that liposomes trigger activation of the NLRP3 inflammasome through mitochondrial reactive oxygen species and calcium influx via the transient receptor potential melastatin 2 channel (14).
During infection, bacterial pathogen escape from the lysosome after phagocytosis is an important signal inducing NLRP3 inflammasome activation (15). However, activation of the NLRP3 inflammasome appears to be dependent on various cell types and stimuli. For example, the NLRP3 inflammasome can be spontaneously activated by primary stimulation of human monocytes, during which stimulation of certain PAMP and DAMP signals provide sufficient signals to trigger mature IL-1β secretion (16). The general concepts of NLRP3 inflammasome activation and the two-signal model are shown in Fig. 1.
Emerging evidence has also revealed involvement of other mechanisms/signaling molecules, beyond the core machinery of the inflammasome complex, in inflammasome activation. A recent study showed that double-stranded RNA-dependent protein kinase (PKR, also known as EIF2AK2) plays an important role in inflammasome activation through physical interaction with several inflammasome components, including NLRP3 (17). Another study showed that guanylate binding protein (GBP) 5, a member of the interferon (IFN)-inducible GBP family, can physically interact with the pyrin domain of NLRP3 and contributes to oligomerization of the NLRP3 inflammasome complex (18). To activate NLRP3 inflammasome, it was suggested that the adaptor ASC on mitochondria should be moved to NLRP3 on the endoplasmic reticulum (19, 20). A recent study proposed a spatial assembly model of microtubule-driven approximation between ASC and NLRP3, which depends on a low intracellular concentration of NAD+ and SIRT2-dependent deacetylation of microtubules (20). The NLRP3 inflammasome-activating signals trigger the recruitment of NLRP3 to mitochondria, and this is dependent on mitochondrial antiviral signaling protein (MAVS) (21), the mitochondrial outer membrane protein that was originally reported as a signaling adaptor leading the production of type 1 interferon (IFN) (22, 23). In addition, voltage-dependent anion channels (VDAC) is essential for NLRP3 inflammasome activation through the uptake of Ca2+ into the mitochondria from MAMs (19). The current model of NLRP3 inflammasome activation and its assembly is shown in Fig. 1.
BACTERIAL INFECTION AND NLRP3 INFLAMMASOME ACTIVATION
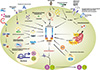
Diverse infectious microbes activate the NLRP3 inflammasome. In this section, we discuss several bacterial pathogens and their products that induce NLRP3 inflammasomes in vitro and in vivo (Fig. 2). Earlier studies showed that gram-positive Staphylococcus aureus or Listeria monocytogenes, but not Salmonella typhimurium or Francisella tularensis, require NLRP3 inflammasome activation (24). In addition, Shigella flexneri can cause NLRP3-dependent necrosis; i.e., pyronecrosis, in macrophages; however, this necrotic death is not caspase-1 and IL-1β dependent (25). Other gram-negative enteropathogens, such as enterohemorrhagic Escherichia coli (EHEC) and Citrobacter rodentium, induce NLRP3 inflammasome activation in bone marrow-derived macrophages in a Toll-IL-1 receptor (TIR)-domain-containing adapter-inducing interferon-β (TRIF)-dependent pathway. Interestingly, TRIF plays an essential role in the activation of caspase-11 in NLRP3 inflammasome activation during gram-negative bacterial infection (26). Gram-negative diplococcic Neisseria gonorrhoeae infection promotes NLRP3 inflammasome activation through activation of cathepsin B and leads to NLRP3-dependent monocytic cell death (27).
Although Mycobacterium tuberculosis (Mtb) resists inflammasome activation (28), the atypical mycobacterium Mycobacterium abscessus can induce the maturation and secretion of IL-1β in human monocyte-derived macrophages (29). However, there are controversial results in Mtb-induced NLRP3 inflammasome activation. In human monocytes/macrophages, the M. tuberculosis protein ESAT-6 was reported to stimulate NLRP3 inflammasome by disrupting cell membranes and allowing of Ag85 to access cytosol (30). Moreover, recent studies have shown that murine microglia primed with conditioned media from cultures of macrophages infected with Mtb induce caspase-1 activation and IL-1β secretion after Mtb stimulation in a NLRP3- and ASC-dependent manner, suggesting a role for microglialeukocyte interactions in the pathogenesis of tuberculous meningitis (31). Interestingly, a recent study has shown that Orientia tsutsugamushi, a pathogen causing scrub typhus, induces ASC inflammasome activation to secrete IL-1β in macrophages; however, this was not dependent on NLRP3, NLRP4 or AIM2 (32). Chlamydia pneumoniae, a common respiratory pathogen in atypical pneumonia, induces maturation and secretion of IL-1β in unprimed bone marrow-derived macrophages during productive infection. C. pneumoniae-dependent caspase-1 activation and IL-1β maturation were dependent on NLRP3/ASC (33).
In addition, a variety of bacterial components induce NLRP3 inflammasome activation through diverse mechanisms. Several bacterial toxins; e.g., nigericin (Streptomyces hygroscopicus), valinomycin (several streptomyces strain), maitotoxin (Gambierdiscus toxicus), aerolysin (Aeromonas hydrophila) and listeriolysin O (L. monocytogenes), trigger the NLRP3 inflammasome pathway through pore formation and plasma membrane disruption, as mentioned above (24, 34).
The β-hemolysin of group B streptococci, a frequent life-threatening systemic inflammation and meningitis, can induce secretion of IL-1β, as well as pro-IL-1β synthesis, in bone marrow-derived dendritic cells. Interestingly, the production of IL-1β and pro-IL-1β can be efficiently blocked by bafilomycin treatment, suggesting that group B streptococci-induced inflammasome activation is dependent on bacterial internalization and phagosomal acidification (35). In addition, the pneumolysin virulence factor of S. pneumoniae can induce the NLRP3 inflammasome, leading to enhanced secretion of cytokines, including IL-17A and IFN-γ (36). Recent studies have also reported that the S. aureus virulence factors α-, β-, and γ-hemolysin can activate the NLRP3 inflammasome during S. aureus pneumonia, although pulmonary injury induced by live S. aureus is not dependent on IL-1β signaling (37). Moreover, S. aureus pore-forming toxin PVL activates the NLRP3 inflammasome and subsequent pyronecrosis (38).
A recent study showed that Td92, a surface protein of Treponema denticola, one of the major periodontopathogens, can induce NLRP3 inflammasome activation in macrophages through a direct interaction with the host cell membrane integrin α5β1 (39). Moreover, the cholera toxin B of Vibrio cholerae can induce non-canonical NLRP3 inflammasome activation in a caspase-11-dependent manner (40). The effector YopJ of Yersinia pestis, the causative agent of plague, also leads to activation of the NLRP3/ASC/capsase-1 inflammasome in macrophages through a mechanism dependent on K+ efflux (41). Bacterial mRNA released from the lysosome can be sensed by NLRP3 inflammasome in the cytosol through detection of a special class of viability-associated PAMPs (42). The lipooligosaccharide structure of N. gonorrhoeae potently activates the NLRP3 signaling system, suggesting that the lipooligosaccharide structure is involved in the pathogenesis of gonococcal infections (27). Accumulated knowledge of bacterial pathogens, their components, and the NLRP3 inflammasome activation will evoke a new avenue for understanding the pathogenesis, and/or developing potential strategies to treat, bacterial infections.
THE PROTECTIVE ROLES OF NLRP3 INFLAMMASOME ACTIVATION IN BACTERIAL INFECTION
Extensive research has revealed that NLRP3 inflammasome activation contributes to host defense against numerous bacterial infections. Mice lacking NLRP3, ASC, or caspase-1 were more susceptible to group B streptococcal infection than wild-type mice, suggesting a critical role for the NLRP3 inflammasome in successful anti-streptococcal responses (35). Streptococcal pneumolysin-induced NLRP3 inflammasome activation contributes to protective immunity through induction of IL-17A and IFN-γ secretion against respiratory infection with S. pneumoniae (36). Using a murine model of melioidosis, previous studies showed that the inflammasome components ASC, caspase-1, and NLRP3 are critical for host defense against lung infection with Burkholderia pseudomallei through synthesis and secretion of IL-1β (43). In addition, mice lacking the inflammasome components Nlrp3, Nlrc4, and caspase-1 exhibited increased susceptibility to Citrobacter rodentium infection, which is similar to human infection with enteropathogenic and enterohemorrhagic E. coli (44). In addition, IL-1β- and IL-18-null mice also had increased bacterial burdens and showed an exacerbated pattern of histopathology (44). Moreover, mice lacking both Nlrp3 and Nlrp4 are highly susceptible to S. typhimurium infection, suggesting the redundant roles of the two NLRs in host innate defense against Salmonella infection (45). However, a recent study has shown that caspase-11-not NLRP3, NLRC4, or ASC-is critical for host defense and enhanced clearance of bacteria using a S. typhimurium mutant strain (sifA), which can aberrantly enter the host cell cytosol (46).
During chronic infections caused by Mtb, inflammasome activation and generation of IL-1β may play a dual role in both innate immune defense and inflammatory pathogenic responses. Although AIM2 inflammasomes appear to be protective against Mtb infection (47), NLRP3 inflammasome activation is associated with the immunopathology of tuberculosis, rather than host defense against Mtb infection (48, 49). In Mycobacterium kansasii infection, NLRP3 inflammasome activation and secreted IL-1β derived from caspase-1 activation play an important role in host defense and restriction of intracellular M. kansasii (50). Moreover, Gbp5-deficient mice are defective in caspase-1 cleavage and maturation of IL-1β/IL-18 in vitro and impaired host defense against listerial infection in vivo (18).
In this context, a recent study demonstrated that a network of immune cell populations spatially organized in lymph nodes can efficiently interact with lymphatic sinus-lining sentinel macrophages, and respond rapidly to inflammasome-induced IL-18 produced by these phagocytes, leading to lymphoid cell IFN-γ secretion, which limits systemic spread of the pathogens (51). Thus, inflammasome activation is important for a local and acute innate immune response to limit deep invasion of the lymph nodes by pathogenic bacteria (51).
THE PATHOGENIC ROLES OF NLRP3 INFLAMMASOME ACTIVATION IN BACTERIAL INFECTION
Although the inflammasome activation plays a key role in limiting the spread of an invading pathogen, it can be deleterious due to tissue damage associated with chronic inflammatory responses. When inflammasome activation functions improperly or is uncontrolled, chronic inflammation may develop into various immune disorders, including Crohn's disease, ulcerative colitis, or arthritis (52). IL-1β and IL-18 are potent proinflammatory cytokines that are required for host defense against diverse infections, and simultaneously are involved in various inflammatory pathologies. In addition to processing IL-1β, inflammasome activation often results in the caspase-1-mediated pyroptotic form of cell death, which leads to bacterial exposure, efficient uptake, and killing by neutrophils (53). However, repeated rounds of pyroptosis can lead to severe inflammatory changes and significant damage to host tissues, particularly in the absence of downstream killing by neutrophils (53). TRIF signaling is essential for NLRP3 inflammasome activation by several gram-negative bacteria, and in detrimental inflammation associated with enteric infections (26).
Earlier studies showed that IL-1 secreted by epithelial cells after Chlamydia trachomatis infection is associated with tissue damage to the Fallopian tube, potentially leading to infertility (54). It was also reported that C. trachomatis infection of human monocytes triggers secretion of IL-1β through NLRP3 inflammasome activation, which induces an inflammatory response to this pathogen (55). In genital infection with Chlamydia muridarum, mice lacking NLRP3, NLRC4, and ASC showed no reduction in oviduct pathology compared with control mice, suggesting a limited role for the inflammasome pathway during genital chlamydial infection (56).
In Helicobacter pylori infection, IL-1β is primarily proinflammatory, whereas IL-18 counteracts the inflammatory responses induced by IL-1β and Th17 (57). Interestingly, two caspase-1 substrates act together to achieve balanced control of Helicobacter infection and avoid excessive gastric pathology (57). The gram-negative spirochete, Borrelia burgdorferi, a pathogen of chronic Lyme disease-associated arthritis, activates the inflammasome in a manner dependent on ASC and caspase-1, but not NLRP3 (58, 59). In addition, IL-1 is a major pathogenic mediator of Lyme arthritis and is not required for control of Lyme borreliosis in murine models (59, 60).
Activation of the NLRC4 inflammasome by the flagella of Pseudomonas aeruginosa is deleterious to bacterial clearance in acute pneumonia and associated with increased cell death (61). In corneal infection with P. aeruginosa, caspase-1 deficiency leads to inhibition of cytokine and chemokine production, and reduced infiltration of polymorphonuclear leukocytes, suppressing corneal damage and inflammation (62). These studies suggest that targeting the inflammasome limits the pathological responses during P. aeruginosa infection.
In Mycobacterium marinum infection, the ESX-1 (type VII) secretion system, a major virulence determinant, promotes NLRP3 and the ASC-dependent inflammasome. However, NLRP3 inflammasome activation by M. marinum is responsible for host-detrimental effects without restricting bacterial growth (48). Similarly, the ESX-1 secretion system and ESAT-6 proteins induce NLRP3 inflammasome activation, which leads to necrotic death of THP-1 human macrophages via a Syk tyrosine kinase (63). In vivo, an Mtb aerosol infection study indicated that neither Nlrp3 nor Casp-1 null mice show any differences in pulmonary bacterial burden or long-term survival, when compared with wild-type mice (64). However, ASC appears to be important in host protection against chronic Mtb infection because ASC-deficient mice exhibit reduced survival and defective granuloma formation (64). Moreover, killed Mtb, the immunostimulatory component in complete Freund's adjuvant, promotes experimental autoimmune encephalomyelitis through activation of the inflammasome complex in dendritic cells and increased IL-17 production by CD4 and γδ T cells (65). These findings suggest that modulation of inflammasome activation may prevent tissue damage and host death in response to a variety of bacterial infections.
MODULATION OF NLRP3 INFLAMMASOME ACTIVATION BY PATHOGENIC BACTERIA
Evidence of manipulation of inflammasome activation by bacterial pathogens has accumulated. Bacteria use diverse strategies to hijack or manipulate inflammasome activation in host cells (66). Several bacterial proteins that modulate NLRP3 inflammasome activation for immune evasion have been described. Earlier studies indicated that P. aeruginosa ExoS, a type III effector protein, negatively regulates caspase-1-dependent IL-1β maturation through a mechanism dependent on its ADP ribosyltransferase activity (67). In addition, Yersinia YopE and YopT prevent the activation of caspase-1 through inhibitory effects on Rac1 (68). The Yersinia type III secreted effector protein, YopK, can inhibit inflammasome activation through interaction with the T3SS translocon (69). Detection of the Yersinia type III secretion system (T3SS) by NLRP3 and NLRC4 induces the assembly of inflammasomes, and promotes bacterial clearance (69); YopK-mediated inhibition of inflammasome activation may have detrimental effects on host defense.
In addition, Legionella pneumophila can manipulate ASC levels to modulate inflammasome and NF-κB pathway activation, thus evading immune evasion and promoting growth (70). When ASC was over-expressed in cells to maintain constant levels of protein during infection, caspase-1 activation and restriction of Legionella growth resulted (70). The zinc metalloprotease Zmp1 of Mtb is involved in the prevention of inflammasome activation by Mtb. Zmp1-deleted Mtb enhances the secretion of IL-1β through inflammasome activation, which plays a role in Mtb phagosomal maturation and enhancement of mycobacterial clearance in murine aerosol-infection models (28). The Francisella tularensis RipA protein inhibits secretion of IL-1β, IL-18, and TNF-α by macrophages to escape from host immunity, and an F. tularensis ripA-deletion mutant (LVSΔripA) induced higher levels of inflammatory cytokines in animals (71). Collectively, these data demonstrate that a class of bacterial components modulates inflammasome recognition and assembly to evade the host immune system and promote bacterial survival within host cells/tissues.
CONCLUSION
NLRP3 inflammasome activation is crucial for recognition of cytosolic pathogenic microbes and allows the host to induce an appropriate protective innate immune response. A two-signal theory of triggering NLRP3 inflammasome assembly and activation has been proposed. However, evidence of activation or regulation of the inflammasome complex by other mechanisms/molecules has accumulated. Interestingly, many bacteria and their products can manipulate and/or activate the NLRP3 inflammasome complex. Future studies will identify additional host molecules or bacterial components that activate the inflammasome pathway.
It is clear that NLRP3 inflammasome activation contributes to host defense during a variety of bacterial infections. However, chronic inflammation accompanied by dysregulated inflammasome activation during persistent infection may result in detrimental effects and immunopathologies. In addition, many pathogenic bacteria subvert the NLRP3 inflammasome pathways, often through inhibition of, or interaction with, the inflammasome core machinery by virulence factors. In future research, additional virulence factors and mechanisms by which successful microbial pathogens modulate host inflammasome pathways will be clarified. Current knowledge of inflammasome activation or regulation by pathogens or their components must be expanded using appropriate genetic and/or animal models. Understanding how pathogens subvert host inflammasome signaling pathways will contribute to development of interventions to control bacterial infection and provide protective innate immunity to the host.
 XML Download
XML Download