

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Multiple endocrine neoplasia type 1 (MEN1) syndrome is an autosomal dominant inherited disorder, which predisposes patients to the development of varying combinations of endocrine and non-endocrine tumors (1). They include the hyperplasia or adenoma of the parathyroid gland, and tumors of the endocrine pancreas and anterior pituitary (2). However, primary aldosteronism is one of the conditions rarely associated with MEN1 (3). It has also been reported that meningiomas are related to MEN1 (4). The candidate gene, MEN1, is located on chromosome 11q13 and loss of heterozygosity (LOH) of the MEN1 locus appears to MEN1-related tumors (5). Here we report a rare case consisting of primary hyperparathyroidism associated with primary aldosteronism, Hürthle-cell thyroid carcinoma, and meningioma.
CASE DESCRIPTION
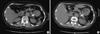
A 68-yr-old woman was referred to the Department of Endocrinology for an elevated serum calcium level (2.95 mM, normal value: 2.25-2.75 mM) on May 24, 2010. Five years earlier, she had visited to the Department of Nephrology for evaluation of hypokalemia. At that time, she had been diagnosed with primary hyperaldosteronism with a low potassium level (1.7 mM, normal value: 3.5-5.3 mM), a high plasma aldosterone level (0.244 nM, normal value: 0.014-0.083 nM), and a low normal plasma renin level (0.021 pM, normal value: 0.008-0.3 pM). Adrenal CT scan showed a 1-cm round nodule in the left adrenal gland (1.1 × 1.0 cm), suggestive of adrenal adenoma (Fig. 1A). At that time, she refused an operation and started to take spironolactone. After 5 months, however, she did not visit the outpatient clinic any more.
Two years before the presentation reported here, she came to the Department of Neurology, complaining of the involuntary movement of her neck and four extremities. She was referred again to the Department of Nephrology with a similarly low potassium level (1.8 mM, normal value: 3.5-5.3 mM), and high plasma aldosterone level (aldosterone 0.327 nM, normal value: 0.014-0.083 nM),and low normal plasma renin level (0.040 pM, normal value: 0.008-0.3 pM). On the follow-up CT scan, the size of the left adrenal adenoma had slightly increased since the previous film (1.4 × 1.2 cm) (Fig. 1B). She started to take 50 mg of spironolactone again. Because of other vague symptoms like depression, anxiety, headache, nausea, anorexia, and polydipsia, it was also recommended that she visit a neuropsychiatric clinic under the suspicion of manic depressive disorder.
When she visited the Department of Endocrinology due to hypercalcemia, she had taken spironolactone for 2 yr, maintaining a normal potassium level. She had been taking antihypertensive medication for 20 yr and hypoglycemic agents for 2 yr. She had no family history of the endocrine tumors such as parathyroid gland, pancreas or pituitary gland. Further studies demonstrated that her hypercalcemia was associated with an elevated intact-PTH level (83.2 ng/L, normal value 14-72 ng/L). An increased parathyroid uptake of MIBI scan was noted in the right superior, right inferior, and left inferior parathyroid glands (Fig. 2). Multiple cystic and solid nodules, suggestive of malignancy, were incidentally found in both thyroid glands by neck sonography and fine needle aspiration cytology (Fig. 3). During the parathyroid exploration, total thyroidectomy was performed at once. The pathological diagnosis of the resected parathyroid glands was chief cell hyperplasia and that of the thyroid gland was Hürthle-cell carcinoma (Fig. 2, 3).
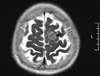
Due to her lasting headache during hospital days, we performed an imaging study. On her brain MRI, a small meningioma (2.1 × 2.0 × 1.4 cm) was detected, as a non-endocrine tumor, in the left parasagital-frontoparietal convexity (Fig. 4); however, there were no pituitary lesions. We decided to follow up on the meningioma without surgery because of the small mass size and no correlating symptoms. Her general condition markedly improved and her adrenal mass and meningioma are now being closely observed.
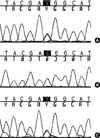
Based on these results, we could conclude that she had primary hyperparathyroidism, primary hyperaldosteronism, Hürthle-cell carcinoma of the thyroid gland, and meningioma. From various clinical manifestations, we suspected that the findings of the patient might have been related to a MEN1 mutation or other genetic background. In the analysis of all coding exons of MEN1 of the patient, polymorphisms were detected in exon 9 and exon 10, but failed to show a germline mutation (Fig. 5A). However, we could find the LOH of the MEN1 locus at codon 1269 (C1269T), in exon 9 of the parathyroid gland, suggesting a MEN1-related tumor (Fig. 5B). In case of the thyroid glands, there was no LOH of MEN1 locus at codon 1269 (Fig. 5C).
DISCUSSION
We report here a rare case consisting of primary hyperparathyroidism associated with primary hyperaldosteronism, Hürthle-cell thyroid carcinoma, and meningioma. We suspected those serial findings might have come from a MEN1 mutation and tested this using genomic DNA from peripheral blood leukocytes. In this patient, we could not find a germline mutation in the protein-coding lesion of the MEN1 gene, but detected the LOH of the MEN1 locus in exon 9 of the parathyroid gland, suggesting a MEN1-related tumor.
A practical definition of MEN1 states that a patient must have abnormalities in at least two of the affected endocrine glands, as well as a first-degree relative with at least one MEN1-related lesion or a known MEN1 mutation (2, 6). This usually involves the parathyroids (95%), pancreas (40%), or pituitary (29%), but occasionally other endocrine glands are affected as well (7, 8). Our patient had a parathyroid lesion, but neither pancreas nor the pituitary lesions. However, it is well known that MEN1 mutation relates to combinations of over 20 different endocrine and non-endocrine tumors (1). Thus, no simple definition of MEN1 could cover all index cases or all families. Some researchers have called those atypical types of patients as 'MEN1 phenocopy' or 'MEN1 phenocopy variants'.
The prevalence of adrenal lesions in MEN1 ranges from 9% to 55% (3, 8), and is generally reported to be rarer than those of parathyroid or pancreatic endocrine lesions (11, 12). Furthermore, "functional" aldosterone-producing adrenocortical adenoma is rarely seen. In earlier studies, some researchers found LOH of the 11q13 locus in aldosterone-producing adrenal adenoma (8, 9). It was notable that primary hyperparathyroidism was newly detected after treating hyperaldosteronism with spironolactone. It has been reported that hypercalcemia might be masked for a while under hyperaldosteronism, which causes hypokalemic alkalosis and a low serum ionized calcium (13).
Thyroid disease can be observed in over 25% of MEN1 patients (12). Even though there have been a few reports of papillary thyroid cancer (14-16), no report has associated Hürthle-cell thyroid cancer with MEN1-like syndrome. In this case, we found Hürthle-cell thyroid cancer incidentally during exploration. Although we could not find somatic changes like LOH of the MEN1 locus in the thyroid tissue, this is the first report of Hürthle-cell thyroid cancer combined with MEN1-like phenotype.
Meningioma was also accompanied by other endocrine tumors in this patient. Asgharian et al. recently reported that meningiomas developed in 6 of 74 (8%) MEN1 patients with pancreatic endocrine tumors (PETs) prospectively. According to them, meningiomas were 11 times more frequent in patients with PETs and MEN1 than without it (4). Bäcklund et al. (17) also studied the relationship between parathyroid adenoma and primary CNS tumors, particularly meningiomas. They reported that the incidence of meningiomas in patients with parathyroid adenoma was much higher than in those without it. Although more research is needed, there might be a shared etiology of parathyroid adenomas and meningiomas.
The MEN1 gene is a tumor suppressor gene on chromosome 11q13 that encodes the nuclear protein, menin, which regulates cell growth and cycle, and when this protein deficit occurs, MEN1 develops (10). In addition, LOH at the locus of the MEN1 tumor suppressor gene may have an important role in the development of tumor (5). In this patient, we found the LOH of the MEN1 locus of the parathyroid gland, but could not detect a germline mutation in the MEN1 gene. There might be several explanations for the lack of definite mutations. First, the sensitivity of the PCR and direct sequencing method is generally 60%-70%. MEN1 germline mutations have been identified in about 80% to 90% of probands with familial MEN1 syndrome (2) and about 65% of individuals with simplex MEN1 syndrome (5). There is no doubt about the usefulness of MEN1 mutation analysis to confirm a diagnosis of MEN1, but we should also note the limitation of genetic analysis indicated above. Next, approximately 10% of new germline mutations are being detected among all MEN1 patients, which is one of the reasons why the association of genotype and phenotype could not be identical in our case. Based on a few recent studies, the incidence of CDKN1B mutations in patients with a MEN1-related phenotype is estimated to be 1.5%-3.7% (18-20).
Our patient has shown more than two endocrine tumors over several years, suggesting MEN1-related clinical features. In this patient, we found the LOH of the MEN1 locus of the parathyroid gland, but could not detect a germline mutation in the MEN1 gene. Considering a variety of phenotypic expression and a limitation of current molecular analysis, periodic screening and follow up will be needed in the patient who have a MEN1-like phenotype but test-negative for MEN1 mutations.


 XML Download
XML Download