

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Renal ischemia and reperfusion (IR) injury is a major cause of acute kidney injury during the perioperative period, and occurs frequently after aortovascular surgery or kidney transplantation (1, 2). The pathophysiology of IR injury includes both direct cellular damage as the result of the ischemic insult as well as delayed dysfunction and damage insulting from activation of inflammatory pathways (3). Tumor necrosis factor (TNF), adenosine, nitric oxide (NO), heat shock proteins (HSP), insulin-like growth factor, and cell adhesion molecules have been implicated in mechanisms that limit IR-induced injury following ischemic preconditioning (IPC) (4). However, the signaling pathway activated by IPC that prevent IR-induced damage is incompletely characterized.
Recent studies have shown that activation of the Akt pathway plays a protective role during IR (3, 5, 6). Also, we previously demonstrated acute and delayed phases of renal protection with renal IPC, and that acute IPC is mediated via G-protein (Gi/o) and Akt phosphorylation, whereas delayed IPC involves Gi/o and inducible nitric oxide synthase (iNOS) upregulation (7).
This study was designed to investigate the hypothesis that reactive oxygen species (ROS) may be released by hepatic IPC as a remote organ and carried to the target tissue in the blood stream, making them available to activate Akt (protein kinase B) signaling pathways and reduce inflammatory reactions. The study of the effects of hepatic IPC on distant kidney may help clarify the protective mechanisms of IPC, which may lead to the identification of new molecular targets for therapeutic interventions.
MATERIALS AND METHODS
Induction of hepatic IPC and renal IR injury
All experiments conformed to the guidelines of and were approved by the Institutional Laboratory Animal Care and the Ethical Committee of the Catholic University of Korea, Seoul, Korea (#2011-0035) on 8 February 2011. All animals were housed in separate cages with a 12 hr light-dark cycle and sufficient water and food. Mice were allowed to acclimatize for 7 days before the experiments. All experiments on mice were performed during the period from March 1 to 31, 2011.
Male C57BL/6 mice (25-30 g) were anesthetized with intraperitoneal pentobarbital, 50 mg/kg or to effect. Mice were placed under a heating lamp and on a 37℃ heating pad. After a midline laparotomy and intraperitoneal application of 20 U of heparin, all mice had a right nephrectomy and dissection of the left renal and hepatic pedicle. The mice were divided into four groups. Group I (sham, n = 8) had a sham operation to assess the effect of laparotomy and dissection of the left renal and hepatic pedicle after right nephrectomy. In group II (IR, n = 8), group III (IPC-IR, n = 8) and group IV (MPG - IPC + IR, n = 8), renal IR injury was created by placing a microvascular clamp on the left renal pedicle for 30 min (1, 7). In group III, hepatic IPC was achieved by brief clamping of the hepatic pedicle for 10 min, followed by 10 min of reperfusion (3), before renal IR injury. In group IV, mice were pretreated with 100 mg/kg N-(2-mercaptopropionyl)-glycine (MPG; a free radical scavenger) 15 min before hepatic IPC and were subjected to renal IR injury (7).
Twenty-four hours after renal IR injury, all mice were re-anesthetized and plasma samples and left kidneys were obtained. For studying the renal effect of hepatic IPC, mice (n = 16) were subjected only to hepatic IPC without renal IR injury, and the left kidney was isolated 15 min (acute, n = 8) or 24 hr (delayed, n = 8) later. The experiments with pretreated MPG mice (n = 16) were performed in the same way.
Immunoblot analyses for protective proteins
The harvested kidneys were quickly frozen using liquid nitrogen and stored at -80℃. They were dissected on ice and immediately placed on ice-cold radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, and 1% Triton-X100 [pH 7.4]) and homogenized for 10 sec on ice. Tissues were centrifuged at 32,500 g and 4℃ under a vacuum for 1 hr. The supernatant was collected and used for immunoblotting as described previously (1, 7). Samples were stabilized with Laemmeli buffer after determining their protein amounts by Bradford assay (BioRad, Hercules, CA, USA) using bovine serum albumin as standard. The proteins, separated by 4 hr 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis at 80 V, were transferred to polyvinylidene fluoride membranes. Membranes were washed with Tris-buffered saline containing 0.1% Tween-20 (TBST) and coated with a blocking solution (5% dry milk in TBST). Measurements of iNOS, phosphorylated Akt (p-Akt), and total Akt were made by immunoblotting. The primary antibodies for p-Akt and total Akt were from Cell Signaling Technologies (Danvers, MA, USA). iNOS antibody was from BD Biosciences Pharmingen (San Jose, CA, USA). The secondary antibody (goat anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase at 1:5,000 dilution) was detected with enhanced chemiluminescence immunoblotting detection reagents (Amersham, Piscataway, NJ, USA), with subsequent exposure to a CCD camera coupled to a UVP Bio-imaging system (UVP, Upland, CA, USA). The band intensities of the immunoblots were within the linear range of exposure for all experiments.
Semiquantitative reverse transcriptase polymerase chain reaction (RT-PCR) for mRNA of proinflammatory cytokines
Semi-quantitative RT-PCR was carried out to analyze the expression of the proinflammatory cytokines (TNF-α) and intercellular adhesion molecule 1 (ICAM-1) (8, 9). A 1 mL aliquot of Trizol reagent (MRC, Cincinnati, OH, USA) was added to the harvested kidneys, which were homogenized and allowed to stand for 5 min. After adding 0.2 mL 1-bromo-3-chloropropane (Sigma-Aldrich, St. Louis, MO, USA), centrifugation was carried out for 15 min at 14,000 g. Subsequently, after separating supernatants and adding an equal amount of phenol: chloroform (5:1; pH 4.7) (Sigma-Aldrich), centrifugation was carried out for 10 min at 14,000 g. After separating supernatants, adding 100% ethanol and allowing the solution react for 1 hr at -20℃, centrifugation was carried out for 10 min at 14,000 g. The solution was kept in at -70℃ after discarding supernatants, washing RNA deposits with 75% ethanol, adding diethylene-pyrocarbonate water, and allowing the solution to react for 5 min at 60℃. RNA concentrations were determined by spectrophotometric absorbance at 260 nm. With extracted RNA (0.5 µg) as the template, cDNA (50 µL) was composed using avian myeloblastosis virus (AMV) reverse transcriptase (Promega, Madison, WI, USA). PCR was done after generating the last reaction liquid 10 µL, including dNTPs (each at 2.5 mM/L), 1 unit I Start Taq DNA polymerase (Intron, Seoul, Korea), and each primer of 20 µM using composed cDNA of 2 µL (Table 1). PCR was done for 35 cycles through 30 sec denaturation at 95℃, 30 sec annealing at 55℃, and 1 min extension at 72℃, and completed after reacting for 10 min at 72℃. β-actin (228 bp) was the internal control gene to determine whether appropriate mRNA had been separated from cells. Extended PCR products were quantified using Gel DocTM XR (BioRad) after 2.0% agarose gel electrophoresis and staining with ethidium bromide dye.
Measurement of serum creatinine level
Renal function was assessed by measurement of serum creatinine 24 hr after renal IR injury. Serum creatinine levels were measured spectrophotometrically using a commercially available quantitative colorimetric assay (Sigma-Aldrich) (10).
Histological examinations
The harvested kidneys were fixed in 10% formalin solution, embedded in paraffin, sectioned at 5 µm thickness, and stained with hematoxylin-eosin. Morphological assessment was performed by an experienced renal pathologist who was unaware of the treatment that the animal had received. A previously described grading scale of 0-4 (11) was used for the histopathological assessment of ischemia and reperfusion-induced damage of the proximal tubules.
Statistical analyses
Data are mean ± SEM. Statistical processing was done using SPSS 12.0 (SPSS, Chicago, IL, USA). Data were analyzed using t test or the analysis of variance (ANOVA). The ordinal values of the Jablonski scale were analyzed by the Kruskal-Wallis non-parametric test with Dunnett's test comparison among groups. A P value of < 0.05 was considered statistically significant.
RESULTS
Immunoblotting for iNOS, p-Akt, and Akt
Kidneys collected 15 min after hepatic IPC displayed elevation of p-Akt by 85% without affecting iNOS or Akt expression (P = 0.011) (Fig. 1). Kidneys collected 24 hr after hepatic IPC displayed a significant elevation (44%) in the expression of iNOS (P = 0.038), but elevated expressions of p-Akt at the acute phase were abolished. On the other hand, elevations of protective proteins caused by hepatic IPC did not appear in acute and delayed phase of pretreated MPG mice (Fig. 1).
RT-PCR of proinflammatory genes
Kidney inflammation was determined by the measurement of mRNA-encoding markers of inflammation, including TNF-α and ICAM-1 at 24 hr after renal IR injury. Although there were significant elevations in TNF-α and ICAM-1 level in group II (66.7 ± 8.7 and 91.9 ± 13.7; P < 0.001, respectively) vs group I (44.6 ± 7.5 and 61.7 ± 8.9, respectively), these elevations were markedly attenuated in group III (52.4 ± 7.4 and 67.3 ± 10.1, respectively; P = 0.003 and P = 0.001) vs group II (Fig. 2). Proinflammatory genes were significantly re-elevated in MPG pretreated group IV (62.6 ± 8.3 and 89.7 ± 12.8, respectively; P = 0.022 and P = 0.002) vs group III (Fig. 2).
Protective effects against renal IR injury of hepatic IPC
As expected, serum creatinine at 24 hr after reperfusion was significantly elevated in group II (IR, 2.7 ± 0.6 mg/dL; n = 8; P < 0.001) and group IV (MPG - IPC + IR, 2.2 ± 0.5 mg/dL; n = 8; P < 0.001) compared with group I (sham operation, 0.4 ± 0.1 mg/dL; n = 8) (Fig. 3). Hepatic IPC in group III (IPC + IR, 0.6 ± 0.1 mg/dL; n = 8; P < 0.001) significantly improved renal function compared with group II. However, renal protective effects of hepatic IPC were lost by MPG treatment in group IV (P < 0.001) vs group III (Fig. 3).
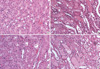
Histopathologic findings at 24 hr after reperfusion were nearly normal in hepatic IPC treated animals (group III) (Fig. 4). In contrast, several areas of tubular dilation and necrosis, medullary luminal congestion, and hemorrhage were present in the kidneys of mice subjected to IR (group II) and pretreated MPG before hepatic IPC and renal IR (group IV) (Fig. 4). Jablonski histology grading scores in the outer medullary of kidneys are shown in Fig. 5. Groups II and IV resulted in severe acute tubular necrosis (grade 3.5 ± 0.5 and 3.3 ± 0.5, respectively; P < 0.001) vs sham grade (0.2 ± 0.3). Group III (grade 1.1 ± 0.6; P < 0.001) showed significantly improved necrosis scores compared with groups II and IV.
DISCUSSION
Acute kidney injury frequently results from renal IR injury and is a major contributor to morbidity and mortality during the perioperative period. Ischemic preconditioning is a phenomenon whereby brief periods of ischemia protect the organ against a more sustained ischemic insult. Previous studies in animal models demonstrated that renal or hepatic ischemic preconditioning protects against more prolonged IR injury (3, 6, 7). IPC also has protective effects on distant IR injured organs (12-14). But, only a few studies have been conducted to ascertain the effect of IPC on a remote organ; the results were equivocal and the mechanism of IPC was unclear. Therefore, the present study was undertaken to determine whether hepatic IPC could protect against renal IR injury, and to further elucidate the role of ROS induced by hepatic IPC in attenuating this injury.
Many endogenous neurotransmitters, peptides, and hormones have been proposed to function in the signal transduction pathways that mediate the cytoprotective effect of IPC. IPC shares many fundamental steps, including activation of G-protein coupled receptors, multiple protein kinases and ATP-sensitive potassium channels (15). Experimental studies have shown that the protective effects of hepatic IPC are triggered by the release of adenosine and NO and the subsequent activation of signal networks involving protein kinases such as phosphatidylinositol 3-kinase (PI3K), protein kinase C, and p38 mitogen-activated protein kinases (MAPK), and transcription factors such as signal transducer and activator of transcription 3, nuclear factor-κB and hypoxia-inducible factor 1 (16). NO modulates mitochondrial function, mediates cytoprotection after IR, and has been implicated in the signaling of the highly protective IPC program (17, 18). In cardiac and neuronal IR injury, acutely protective effects of IPC dissipate over several hours but reappear 24-72 hr later. This phenomenon is defined as a second window of protection, or delayed IPC, and is proposed to be mediated by iNOS upregulation and enhanced release of NO in these organs (7, 19). In this study, we observed approximately 40% upregulation of iNOS in the kidneys of mice 24 hr after hepatic IPC. Inhibition of ROS function with MPG prevented the elevation of iNOS 24 hr after hepatic IPC. Finally, induction of iNOS was critical for the mechanism of delayed renal protection.
The PI3K kinase/Akt pathway is an important survival pathway in protection against IR injury. Activation of this pathway promotes cell survival in part by its modulation of various downstream elements involved in apoptosis. Recent studies have shown that activation of the Akt pathway plays a protective role during IR injury (5, 6). Phosphorylation of Akt caused by IPC begins as early as 15 min after reperfusion (3). Presently, p-Akt also increased by approximately 80% in only 15 min after hepatic IPC, and remained elevated for 24 hr. However, the level of total Akt expression did not significantly change after reperfusion. The major effect in the acute stage after hepatic IPC was an immediate increase in Akt phosphorylation of distant kidney following reperfusion.
Previous animal studies demonstrated that, although high concentrations of oxygen free radicals induce tissue injury during the reperfusion period after prolonged ischemia, moderate oxygen free radicals are an important prerequisite of IPC in cardiac, hepatic, and renal cells (7, 20, 21). Oxygen free radicals phosphorylate several important cytoprotective kinases, including extracellular signal-regulated kinase (ERK1/2), MAPK and Akt, and are involved in the upregulation of several cytoprotective genes (22). ROS are attractive signaling candidates to account for preconditioning in the kidney because renal cells are subject to obligatory bursts of oxidant stress during the reperfusion phase after each preconditioning stimulus. Therefore, distant renal exposure through the blood stream of moderate ROS caused by hepatic IPC may also initiate cytoprotective signaling to defend against subsequent and more severe free radical-mediated injury in renal tubule cells. The present observation that the free radical scavenger MPG prevented the remote renal protective effects by hepatic IPC supports this scenario.
Activation of the Akt signaling pathway and increased NO suppress inflammatory reactions, which are important contributors to renal IR injury (6, 23, 24). NO also regulates neutrophil recruitment by inhibiting the expression of adhesion molecules in the vascular endothelium, resulting in increased blood flow to ischemic regions (25). Therefore, reduction of the inflammatory response by hepatic IPC can produce beneficial effects in renal IR injury. The present study demonstrates that the levels of the proinflammatory cytokines TNF-α and ICAM-1 24 hr after renal IR injury were lower in mice treated with hepatic IPC (group III) than in mice not similarly-treated (group II) and in MPG-pretreated mice (group IV). Attenuation of the expression of these cytokines from distant kidneys by hepatic IPC may contribute to the reduction in renal inflammatory injury after IR. The lesser injury on histopathologic and biochemical studies in kidneys of hepatic IPC pretreated mice was predictive of better renal function.
In conclusion, hepatic IPC directly reduces necrosis and inflammation in distant kidneys via mechanisms involving reduction in proinflammatory mRNAs. Hepatic IPC also induces phosphorylation of Akt and upregulation of iNOS in the kidney through the blood stream of hepatic-released ROS. Clinical manipulation of the signaling pathways of remote IPC that mediate protection may lead to therapeutic improvements to prevent or reduce the incidence of acute kidney injury during the perioperative period.




 XML Download
XML Download