

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
C1qTNF-related protein 6 (CTRP6) was initially identified as a member of adiponectin paralogs (1). It consists of an N-terminal signal peptide, a collagen repeat, and a C-terminal globular domain, and it is widely expressed in many tissues. We previously demonstrated that CTRP6 is involved in fatty acid metabolism in muscle cells by activating AMP-activated protein kinase (AMPK) (2). Recently, we showed that CTRP6 and its globular domain induce IL-10 expression in macrophages (3). However, the regulatory mechanism of CTRP6 expression remains largely unknown.
Mitochondrial dysfunction has been implicated in several metabolic diseases, such as obesity, hyperlipidemia, and diabetes (4). We previously showed that depletion of mtDNA in muscle cells decreases the expression of insulin receptor substrate-1 (IRS-1), thereby causing impaired glucose utilization and insulin resistance (5). Mitochondria-to-nucleus stress signaling results in a variety of changes in nuclear gene expression (6-8). A recent study observed that CTRP6 expression is increased in adipose tissue of 8-week-old and 12-week-old ob/ob mice (1). The activity of mitochondrial respiratory chain complexes in ob/ob mice is between 30% to 50% lower compared to that in wild-type animals (9). Our recent study showed that mtDNA depletion increases the expression of C1qTNF-related protein 5 (CTRP5) gene (10), which shares structural and functional properties with CTRP6. Thus, we investigated the change in CTRP6 expression in response to mtDNA depletion. In this study, we provide evidence that depletion of mtDNA up-regulated CTRP6 expression via an increase in mRNA stability.
MATERIALS AND METHODS
Reagents and antibodies
Polyclonal anti-CTRP6 antibody was purchased from Abcam (Cambridge, MA, USA). Polyclonal anti-actin antibody was obtained from Sigma (St. Louis, MO, USA). Horseradish peroxidase (HRP)-conjugated anti-mouse IgG and anti-rabbit IgG were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Oligonucleotide primers were purchased from Bionics (Seoul, Korea). Sodium pyruvate, uridine, and actinomycin D were obtained from Sigma.
Cell culture
C2C12 cells were maintained in DMEM (high glucose) medium supplemented with 10% (v/v) fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA) and appropriate antibiotics. Depletion of mtDNA was induced by treatment with ethidium bromide (EtBr, 0.2 µg/mL) for 2-3 weeks. EtBr-treated C2C12 cells were grown in the presence of 1 mM sodium pyruvate and 50 µg/mL uridine, which have been shown to be essential for the growth of cells lacking mtDNA (11). Control C2C12 cells were maintained for the same time period under normal culture conditions. Removal of EtBr from the medium restored the mtDNA content to a level similar to that of control C2C12 cells within 4 weeks.
Genomic DNA extraction and polymerase chain reaction (PCR)
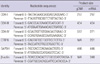
Total genomic DNA was isolated from control, mtDNA-depleted, and reverted C2C12 cells using a DNeasy Tissue kit in accordance with the manufacturer's instructions (Qiagen, Valencia, CA, USA). The amplification of mtDNA was performed under the following conditions: 94℃ for 2 min (initial denaturation); 94℃ for 30 sec, 55℃ for 30 sec, 72℃ for 30 sec (25 cycles for COX-III and GAPDH; 30 cycles for COX-I and COX-IV); and 72℃ for 10 min (final extension). The primers used in this study are shown in Table 1.
RNA extraction, RT-PCR, and quantitative real-time PCR
Total RNA was extracted from control, mtDNA-depleted, and reverted C2C12 cells using Trizol reagent in accordance with the manufacturer's instructions (Invitrogen). The reverse transcription reaction was performed using M-MLV reverse transcriptase according to the manufacturer's instructions (Promega, Madison, WI, USA). The amplification of CTRP6 was performed under the following conditions: 94℃ for 2 min (initial denaturation); 94℃ for 30 sec, 55℃ for 30 sec, 72℃ for 45 sec (30 cycles for CTRP6; 25 cycles for β-actin); and 72℃ for 10 min (final extension). The primers used in this study are shown in Table 1. For real-time PCR, the resulting cDNA was diluted 2.5-fold and used to amplify CTRP6 or β-actin as a control. Real-time PCR amplification was carried out using SYBR green master mix (Roche Applied Science) in a LightCycler 480 (Roche Applied Science, Mannheim, Germany) as follows: initial denaturation at 95℃ for 5 min; 45 cycles of amplification with denaturation at 95℃ for 30 sec, annealing at 58℃ for 30 sec, and extension at 72℃ for 30 sec; 1 cycle of melting curves at 95℃ for 5 sec, 65℃ for 1 min, and 97℃ continuous; and a final cooling step at 40℃ for 30 sec. The comparative cycle threshold (CT) method was used to analyze the data by generating relative values of the amount of target cDNA as previously described (5).
Measurement of cellular ATP levels
Cellular ATP levels were measured using a Somatic cell ATP assay kit (Sigma, St. Louis, MO, USA) as previously described (12). Briefly, cells were lysed with an ATP releasing solution (Sigma), and the lysates were assayed for luciferase activity in accordance with the manufacturer's instructions (Sigma). ATP-dependent formation of light was measured in a Luminometer (Berthold Technologies, Bad Wildbad, Germany) with appropriate ATP standards.
Western blotting
Equal amounts (20 µg) of total cell lysates were subjected to SDS-PAGE on 10% resolving gels as previously described (13). Separated proteins were electrophoretically transferred to a nitrocellulose membrane (Bio-Rad, Philadelphia, PA, USA). The membrane was blocked with 5% skim milk in Tris-buffered saline (TBS), followed by incubation with anti-CTRP6 or anti-actin antibody overnight at 4℃. After washing with TTBS (TBS with 0.1% Tween 20), the membrane was incubated with HRP-conjugated anti-mouse or anti-rabbit IgG for 1 hr at room temperature. Blots were developed using an enhanced chemiluminescence (ECL) kit (Amersham, Buckinghamshire, UK).
Dual luciferase reporter assay
The mouse CTRP6 promoter region (pCTRP6-1064/+126) was amplified from genomic DNA of C2C12 cells by PCR using the following primers: forward primer, 5'-AAA AGC TAC CCT CGT TCC ACC CCT AGT C-3'; reverse primer, 5'-AAA ACT CGA GCC TGC GGC CTC AAC GTG AAC-3'. The PCR product was cloned into the NheI-XhoI sites of the pGL3/basic vector (Promega, Madison, WI, USA). Control, mtDNA-depleted, and reverted C2C12 cells in a 6-well plate were cotransfected with pCTRP6-luc vector together with pRL-SV40 for constitutive expression of Renilla luciferase as an internal control. Luciferase activity was measured 48 hr after transfection using a Dual Luciferase assay kit (Promega, Madison, WI, USA). The ratio of firefly luciferase activity to Renilla luciferase activity was presented as fold induction of that observed in control C2C12 cells.
mRNA stability analysis
Control, mtDNA-depleted, and reverted C2C12 cells were incubated with 12.5 µg/mL actinomycin D to inhibit transcription. The cells were harvested at six different time points following actinomycin addition, after which total RNA was isolated as described above. The level of CTRP6 transcript was analyzed by RT-PCR and quantitative real-time PCR.
RESULTS
Establishment and characterization of mtDNA-depleted C2C12 cells
To generate partially mtDNA-depleted muscle cell lines, we treated C2C12 cells with EtBr in medium containing pyruvate and uridine, which are known to be essential for the growth of mtDNA-depleted cells (11). The mtDNA content of the EtBr-treated C2C12 cells was measured by amplifying genomic DNA. As shown in Fig. 1A, cytochrome oxidase subunits I (COX-I) and III (COX-III), which are encoded only in mtDNA, were rarely amplified from the genomic DNA of the EtBr-treated C2C12 cells compared to the control C2C12 cells. Removal of EtBr from the culture medium (reverted cells) restored the mtDNA content of the mtDNA-depleted cells to a level similar to that of control C2C12 cells (Fig. 1A). In contrast, nuclear DNA-encoded COX-IV was equally detected in control, mtDNA-depleted, and reverted cells (Fig. 1A). We next analyzed the differential expression of COX-I, -III, and -IV genes in the control, mtDNA-depleted, and reverted cells using quantitative real-time PCR. As shown in Fig. 1B, the mRNA contents of COX-I and COX-III were significantly reduced in the mtDNA-depleted cells, whereas the mRNA content of COX-IV remained unchanged, indicating that mitochondrial mRNA levels correlated with cellular mtDNA contents in C2C12 cells. In addition, the mtDNA-depleted C2C12 cells showed a substantial decrease in total cellular ATP compared to control and reverted C2C12 cells (Fig. 1C).
CTRP6 in mtDNA-depleted C2C12 cells
It was previously reported that CTRP5 is increased in mtDNA-depleted myocytes and activates AMP-activated protein kinase (AMPK) (10). Recently, we found that CTRP6 is also involved in fatty acid metabolism via AMPK activation (2). To investigate the change in CTRP6 expression in response to mtDNA depletion, we measured the expression levels of CTRP6 mRNA in control, mtDNA-depleted, and reverted C2C12 cells using quantitative real-time PCR. Expression of CTRP6 mRNA was up-regulated in mtDNA-depleted cells by 2.5-fold compared with control C2C12 cells (Fig. 2A). In agreement with the results of the mRNA analysis, the protein expression level of CTRP6 also significantly increased in mtDNA-depleted C2C12 cells compared to control C2C12 cells (Fig. 2B). Replacement of mtDNA restored both mRNA and protein expression of CTRP6 to levels resembling those in control C2C12 cells (Fig. 2). These results indicate that mtDNA depletion induced up-regulation of CTRP6 expression.
CTRP6 expression and mRNA stability
To determine whether or not the increase in CTRP6 mRNA level was due to transcriptional activation, we generated a mouse CTRP6 promoter fragment upstream of the luciferase reporter gene. However, the activity of the mouse CTRP6 promoter was the same among control, mtDNA-depleted, and reverted C2C12 cells (Fig. 3). Since the increase in steady-state CTRP6 mRNA level was evidently not due to transcriptional activation, we investigated whether or not the rate of mRNA decay might change under mtDNA-depleted conditions. To analyze CTRP6 mRNA decay in control, mtDNA-depleted, and reverted C2C12 cells, we treated cells with actinomycin D, a transcription inhibitor, and examined CTRP6 mRNA levels for the indicated times. As shown in Fig. 4A, decay of CTRP6 mRNA was inhibited in mtDNA-deleted cells compared with control and reverted C2C12 cells. In contrast, the mRNA level of a long-lived message (beta-actin) was unchanged in control, mtDNA-depleted, and reverted cells. The half-life of CTRP6 mRNA increased from approximately 2 hr in normal C2C12 cells to longer than 8 hr in the mtDNA-depleted C2C12 cells (Fig. 4B). Furthermore, replacement of mtDNA restored CTRP6 mRNA decay to a level similar to that in control C2C12 cells. Thus, up-regulation of CTRP6 mRNA by mtDNA depletion was due to an increase in mRNA stability.
DISCUSSION
Long-term treatment with low dose EtBr specifically inhibits replication and transcription of mtDNA without affecting nuclear DNA replication and transcription (14-16), and it has been a useful tool for the investigation of various cellular changes in response to mtDNA depletion (12, 17, 18). Mitochondrial dysfunction induces mitochondria-to-nucleus stress signaling, which changes the expression levels of various nuclear-encoded genes (17-19). In the present study, we generated C2C12 cells containing partially depleted mtDNA (less than 80% of control cells) due to EtBr treatment and showed that depletion of mtDNA led to the increased expression of both CTRP6 mRNA and protein. However, the activity of mouse CTRP6 promoter was not increased in mtDNA-depleted cells compared to control cells. In contrast, depletion of mtDNA increased the promoter activity of CTRP5 gene (unpublished results), which is known to increase its expression in mtDNA-depleted cells (10) and share structural and functional properties with CTRP6. This finding indicates that depletion of mtDNA up-regulated CTRP5 and CTRP6 expression via different mechanisms. Previous studies have reported that mitochondrial dysfunction can modulate gene expression via an increase in mRNA stability (20, 21). In this study, we found that up-regulation of CTRP6 gene in mtDNA-depleted cells was due to an increase in mRNA stability. Our recent study showed that the induction of micro RNA by mitochondrial dysfunction regulates IRS-1 expression at the translational level (22). Thus, it is possible that translational regulation is also involved in the up-regulation of CTRP6 in mtDNA-depleted cells. Further studies are needed to elucidate whether or not CTRP6 expression is regulated at the translational level under mtDNA-depleted conditions.
AMP-activated protein kinase (AMPK) plays an important role in maintaining energy balance in the cells and contributes to protect the body from metabolic diseases such as type 2 diabetes and obesity (23). High plasma fatty acid concentrations interfere with insulin-stimulated glucose transport and are closely associated with insulin-resistant states including obesity and type 2 diabetes (24). Adiponectin improves insulin sensitivity on skeletal muscle by activating AMPK (25, 26). Recently, we demonstrated that CTRP6 is involved in fatty acid metabolism in muscle cells via activation of AMP-activated protein kinase (AMPK), similar to adiponectin function (2). This finding suggests the therapeutic potential of CTRP6 as a metabolic regulator in metabolic diseases such as diabetes and obesity. Although both adiponectin and CTRP6 stimulate fatty acid oxidation via AMPK activation, the serum level of CTRP6 is significantly increased in adiponectin-null mice (1). In agreement with this finding, CTRP6 expression in adipose tissue is increased in 12-week-old ob/ob mice, which express lower levels of adiponectin than control mice (1). It was reported that deletion of mtDNA induces the AMPK energy stress pathway (27). Oxidation of both palmitic and lignoceric acids is significantly increased in the liver of ob/ob mice with low mitochondrial respiratory chain activity (9). However, impartment of mitochondrial function decreases adiponectin synthesis (28). In this study, we observed that CTRP6 expression was increased by depletion of mtDNA. Thus, it is possible that CTRP6 plays a role in compensating for the decrease in adiponectin expression under mitochondrial dysfunction.
In summary, the results in the present study show that depletion of mtDNA increases CTRP6 gene expression in C2C12 cells. Furthermore, it is demonstrated that increased CTRP6 expression in mtDNA-depleted cells can be attributed to an increase in mRNA stability. Considering that CTRP6 is involved in fatty acid metabolism, our identification helps elucidate the molecular mechanism for the control of energy homeostasis in mtDNA-depleted cells.




 XML Download
XML Download