

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Dysferlinopathy is an autosomal recessive disease caused by a DYSF (MIM*603009) gene mutation, located on chromosome 2p13 (1). Dysferlin encoded by the DYSF gene is a 230 kDa protein with seven C2 domains and a single transmembrane domain at the C terminus. Dysferlin is homologous to the Caenorhabditis elegans spermatogenesis factor fer-1 protein that mediates fusion of intracellular vesicles to the sperm plasma membrane. It was suggested that dysferlin might be a vesicle-associated membrane protein involved in docking and fusion of vesicles in muscle cells (2). Therefore, dysferlin has multiple roles in the process of membrane repair, myoblast differentiation, T tubulogenesis and muscle regeneration (3-5). Although dysferlinopathy is caused by a single DYSF gene, it is well-known that dysferlinopathy has various clinical presentations such as distal Miyoshi myopathy (MM), limb girdle muscular dystrophy 2B (LGMD2B), mixed proximodistal (PD), distal anterior compartment myopathy (DACM) and asymptomatic groups (1, 6, 7). This heterogeneity was observed even in the same family with the same mutation (8, 9). It is well-known that LGMD2B is one of the most common forms of limb girdle muscular dystrophy (10, 11). However, there are few reports of patients with dysferlinopathy in Korea (12-14). To characterize the clinical spectrum of Korean dysferlinopathy, we investigated clinical, pathological, laboratory, and radiological features of dysferlinopathy exhibited by Korean patients.
MATERIALS AND METHODS
Subjects
For this study, we reviewed muscle specimens referred to Gangnam Severance Hospital from March 2004 to June 2011. Each muscle specimen was obtained from the patients with suspected myopathy. We found 33 cases with complete or nearly complete dysferlin protein loss in the muscle specimens by immunohistochemistry (Fig. 1A, B). Two patients were withheld due to incomplete medical records. Finally, 31 patients were enrolled for this study. We studied clinical, laboratory, pathologic and radiologic features of each patient. Clinical data included sex, age of symptom onset, disease duration, initial pattern of muscle involvement, family history, and distribution of weakness at diagnosis. Muscle involvement was evaluated clinically using medical research council scales. Laboratory findings included creatine kinase (CK) levels, needle electromyography, and electrocardiography. The serum CK levels were expressed as ×-fold the upper limit of normal values according to the local reference range. Radiological findings were included muscle CT or MRI scans. Muscle specimens were taken from the following muscles: the vastus lateralis (n = 12), biceps brachii (n = 9), deltoid (n = 1), and unspecified muscles (n = 9). The frozen muscle sections (5 µm thickness) from all muscle specimens were stained with routine histopathological stains such as hematoxylin and eosin (H&E), modified Gomori trichrome (modified GT), and nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) stains.
Immunohistochemistry of muscle specimens
The tissues were processed for immunohistochemistry as follows: 5-µm transverse serial sections were obtained from all muscle specimens. Sections were fixed in acetone at 4℃ for 10 min, rinsed in 50 mM Tris-buffered saline (pH 7.5) for 20 min, and incubated for 30 min with a blocking solution containing 2% bovine serum albumin and 5% normal goat serum as described (15). The sections were then incubated overnight in a humid chamber at 4℃ with one of the following antibodies: C-terminal of dystrophin (Leica Microsystems, Newcastle upon Tyne, UK), rod domain of dystrophin (Leica Microsystems), N-terminal of dystrophin (Leica Microsystems), dysferlin (Leica Microsystems), α-sarcoglycan (Leica Microsystems), β-sarcoglycan (Leica Microsystems), γ-sarcoglycan (Leica Microsystems), and δ-sarcoglycan (Leica Microsystems), α-dystroglycan (Millipore, Billerica, MA, USA), and caveolin (BD Biosciences, San Diego, CA, USA). After washing for 20 min in Tris-buffered saline, the sections were incubated with biotinylated goat anti-mouse IgG (Vector Laboratories Inc., Burlingame, CA, USA) for 30 min at room temperature followed by detection with streptavidin-biotin complex immunoperoxidase (VECTASTAIN Elite ABC kit; Vector Laboratories Inc.). Then the slides were developed with 3, 3'-diaminobenzidine (DAB substrate kit for peroxidase; Vector Laboratories Inc.) substrate for 1-5 min. After being washed, the slides were mounted in mounting medium (VectaMount; Vector Laboratories Inc.).
Statistical analysis
The chi-square test and the Fischer's exact tests were used to compare discrete variables, and the independent t-test was used to compare the means of two samples for continuous variables. The relationship between serum CK levels and age at diagnosis or disease duration was assessed using Pearson correlation coefficients. Differences were considered statistically significant at P < 0.05. Statistical analyses were performed with SPSS (version 17.0).
RESULTS
Demographic and clinical characteristics
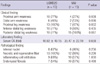
In the study, 12 (38.9%) men and 19 (61.3%) women were included. The mean age of symptom onset was 22.23 ± 7.34 yr (range, 12-36 yr), and 22 patients of 30 symptomatic patients (73.3%) experienced their first symptoms between 15 and 30 yr of age. The mean disease duration was 7.97 ± 6.94 yr (range, 1-30 yr). Table 1 summarizes the clinical and laboratory findings. Three patients had family history of myopathy. Twenty nine patients showed slowly progressive muscle weakness. However, one patient had become symptomatic rapidly during military training. One patient had only extremely high serum CK levels without weakness. Seven patients (23%) described very active and sporty life before symptom onset. Seven patients (two LGMD2B, four MM and one PD patients) had initially asymmetric muscle weakness. Muscle assessment at diagnosis revealed relatively preserved muscle function in the upper limbs.
Laboratory findings
The mean serum creatine kinase (CK) level was elevated 42-fold (min; 4, max; 131) above the upper limit of normal values. There was significant negative correlation between the serum CK and the age of patients (R = -0.652, P < 0.001). However, there was no significant correlation between the serum CK level and the disease duration (P = 0.123). Nine patients recalled that they had had persistent elevation of liver function tests before the onset of weakness and one patient (patient 29) had even received a liver biopsy. Needle electromyography in all patients showed short duration, low amplitude motor unit potentials with full interference patterns. Nine patients had undergone electrocardiograms and all showed normal findings.
Pathological findings
Table 2 shows pathological findings of muscle specimens. The muscle specimens on routine histopathological stains (H&E, modified GT and NADH-TR stains) showed nonspecific dystrophic features of varying degrees with regard to fiber size variability, increased endomysial fibrosis, and necrotic and regenerating muscle fibers. One muscle specimen presented lobulated fibers. However, routine histopathological staining of three muscle specimens did not reveal any pathological changes. Inflammatory cells were observed in the majority of the muscle biopsies and were located especially in the perivascular area (Fig. 1C, D). In the present study, four patients had been misdiagnosed with inflammatory myopathy after muscle biopsy and were treated by steroids and immunomodulating drugs. They could have been properly diagnosed with immunohistochemistry for dysferlin. By immunohistochemistry, all muscle specimens revealed normal staining pattern for other sarcolemmal proteins such as dystrophin, sarcoglycans, dystroglycan and caveolin.
Radiological findings
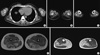
Muscle CT scans were performed in four patients (two LGMD2B and two MM patients). CT scans identified the extent of replacement of skeletal muscle by fat or fibrotic tissue. The upper limb muscles are relatively spared and the lateral posterior compartments of the lower limb muscles had dominantly fatty atrophic changes (Fig. 2A). Muscle MRI scans of the lower limbs were performed in four patients (one LGMD2B and three MM patients). MRI scans revealed that the gastrocnemius, soleus, adductor magnus, hamstrings and vastus lateralis muscles had dominantly fatty atrophic changes and diffuse patchy edema (Fig. 2B).
Comparison between MM and LGMD2B types
We classified 30 symptomatic patients according to their initial patterns of muscle weakness. Fifteen patients had initial clinical presentation of calf weakness in one or both legs (MM group). Thirteen patients had proximal leg weakness (LGMD2B group), and two patients showed both proximal and distal weakness of the leg simultaneously (PD group). There was no difference between the LGMD2B and MM groups in the age of onset (LGMD2B group: 23.69 ± 8.99 yr; MM group: 22.00 ± 6.16 yr; P = 0.562) and the mean disease duration (LGMD2B group: 10.23 ± 6.02 yr; MM group: 7.20 ± 7.74 yr; P = 0.264). There was a tendency for a higher proportion of women in the LGMD2B group (11/13 patients, 85%) than in the MM group (7/16 patients, 47%; P = 0.055). Table 3 shows the difference of clinical and laboratory characteristics between the LGMD2B and MM groups. The weakness of proximal arm and leg is more frequently present in the LGMD2B group than the MM group. Anterior compartment muscles of the lower leg were frequently affected in both LGMD2B and MM groups. In pathological findings and serum CK levels, there were no differences between LGMD2B and MM groups.
DISCUSSION
Our study reveals that clinical and pathological features of Korean dysferlinopathy patients vary greatly. Even though the majority experienced their first symptoms between the ages of 15 and 30, the patients had a broad range of age of onset from 12 to 36 yr. Although classical MM and LGMD2B phenotypes are most common, 31 patients evaluated in this study had a heterogeneous clinical spectrum from isolated hyperCKemia to proximodistal phenotype as shown in previous studies (7, 16-18). The factors responsible for distinct clinical features are yet unknown in our study. Muscle assessment and radiological studies demonstrated that the upper limb muscles were relatively preserved, but posterolateral compartments of the lower limb muscles were dominantly impaired. In the comparison between the MM and LGMD2B groups in our study, there were no differences of onset age, serum CK levels, and pathological findings. The dysferlinopathy patients had markedly elevated serum CK levels. In addition, the serum CK level correlated with the patient's age, but did not correlate with disease duration in contrast with previous studies (7, 19). This is probably associated with a poor relationship with disease progression and the serum CK or histopathological changes of each patient (20). The pathological findings of dysferlinopathy also revealed various features from normal findings to severe dystrophic changes in routine histopathologic stains as were shown in previous studies (21). In our study, four patients had been misdiagnosed with an inflammatory myopathy similar to other reports (22-24). Dysferlinopathy is potentially misdiagnosed as inflammatory myopathy due to several reasons. First, inflammatory cell infiltration is frequently observed in muscle specimens of dysferlinopathy as well as inflammatory myopathy. Inflammatory cell infiltration may be associated with impaired secretion of cytokines and prolonged release of endogenous molecules such as heat shock proteins, high mobility group box 1, and adenosine triphosphates due to compromised membrane repair (25-27). Second, dyferlinopathy and inflammatory myopathy have common features such as very high serum CK levels and young adult onset. Third, many patients with dysferlinopathy had good muscle strength prior to the onset of symptoms (18), which can be suggestive of an acquired myopathy. Recent study revealed the analyzing inflammatory cells, MHC class I expressions and MAC deposits in a muscle specimen may contribute to differentiating dysferlinopathy from inflammatory myopathy (28). However, the best way for differential diagnosis between dysferlinopathy and inflammatory myopathy is to confirm the loss of dysferlin protein or gene.
Dysferlinopathy usually has young adult onset and shows high serum CK levels. However, heterogeneity of clinical presentations and pathological findings on routine histopathological stains makes it difficult to diagnose dysferlinopathy. Currently the most important and easy method for differential diagnosis of dysferlinopathy is immunohistochemical staining. Even though western blotting can quantitatively measure the amount of protein present, it is time-consuming and not compulsory for all proteins on a routine basis. Western blotting can detect the presence of a protein, not the protein function (11). In addition, immunohistochemistry and western blotting both require assessment by invasive muscle biopsy. Even if genetic testing is a less invasive and more accurate method, it is currently limited for diagnosing dysferlinopathy in clinical use due to the large size of dysferlin gene and the absence of a mutational hot spot (29, 30).
In conclusion, limitations in diagnosis by clinical and pathological means currently make immunohistochemistry the most important method for patients suspected to have dysferlinopathy. However, with advancing technology, genetic testing will be the most useful method for diagnosis of dysferlinopathy in the near future.



 XML Download
XML Download