

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hereditary sclerosing poikiloderma (HSP) is a very rare autosomal dominant genetic disease which was first described by Weary et al. (1) in 1969. Clinical manifestations consist of 1) widespread poikiloderma with accentuation in flexural areas and over extensor bony prominences, 2) linear or reticular shaped hyperkeratotic and sclerotic bands on the axillae, antecubital and popliteal fossae, 3) sclerosis of the palms and soles, 4) clubbing fingers, and 5) calcinosis of the tissues. HSP may be also accompanied with musculoskeletal and cardiovascular abnormalities. Therefore, it is necessary to evaluate the existence of other combined abnormalities.
Few cases of HSP have been reported in medical literature, but no Korean case of HSP has been reported yet. Here, we report the first Korean case of HSP presented with poikiloderma and sclerotic bands on both popliteal and antecubital fossae.
CASE DESCRIPTION
A 18-yr-old Korean male visited the Department of Dermatology of the Yeungnam University Hospital due to progressively generalized pigmented lesions on November 2, 2005. He was normal at birth. Reticular hyperpigmented lesions were first observed at 2-yr-old on the popliteal and antecubital fossae. Similar changes gradually became on the axillae and trunk. Linear sclerosing bands appeared on both of the antecubital and popliteal fossae after yr. Most lesions had been progressively increased in size and number with time, but he was asymptomatic.
In family history, his father had similar symptoms and died in his fourties due to cardiovascular disease. Also, his younger brother had similar poikilodermatous lesions. The lesions appeared at the age of 3 yr and hyperpigmented and sclerotic changes had been progressed.
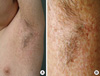
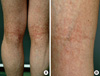
Physical examination revealed micrognathia (Fig. 1A), clubbing fingers (Fig. 1B), and slim extremities compared with trunk (Fig. 1C). Dermatologic examination showed generalized hyperpigmented, telangiectatic, and atrophic changes with accentuation on both axillae (Fig. 2), antecubital, and popliteal fossae. Linear hyperkeratotic and sclerotic bands were found on the both antecubital and popliteal fossae (Fig. 3). Sclerosis of palms and soles was observed.
Laboratory tests, including complete blood cell count, liver function test, blood coagulation test, and antinuclear antibody, and urinalysis were within normal limits or negative. Skin biopsy was performed on the poikilodermatous lesion. Histopathologic finding revealed increased melanin pigments in the basal layer and sclerotic changes and telangiectasia in the upper dermis (Fig. 4A). The fragmentations of elastic fibers in the dermis were noted in elastic stain (Fig. 4B) consistent with a diagnosis of HSP. Cardiac evaluations included electrocardiography and echocardiogram were within normal limits.
After diagnosed as a case of HSP, we decided to observe him without any specific treatment, there was no interval change in his skin for 5 yr. He received regular cardiac evaluation, it revealed no evidence of cardiac disease until now.
DISCUSSION
The diagnosis of HSP is based on the clinical findings and the family history, because no specific tests for the diagnosis have been established (2). The five major clinical manifestations of HSP were proposed by Weary et al. (1). Of these, the most important features for diagnosis for HSP are widespread poikiloderma and sclerotic bands. Previously reported cases suggest that the diagnosis for HSP requires the presence of the above two features and other clinical features are not essential to diagnosis (3). Our patient had generalized poikiloderma, linear sclerotic bands in flexural areas, sclerosis of the palms and soles, and clubbing of the fingers as other reported cases, but tissue calcinosis was not found. In addition, micrognathia was noted. Also, his height is 155 cm and body weight 46 kg, these are similar to an average of sixth grade of elementary school in Korean. Therefore, His growth had lagged behind the industry at least six yr. Thus, our patient was able to diagnose as a HSP.
The widespread poikiloderma characterized by speckled hypopigmentation and hyperpigmentation, slight atrophy, and minimal telangiectasia mainly involves in the flexural areas and over bony prominences (2). It is absent at birth and first appears within the second to fourth yr of life without prior vesiculation or eczema and progressively worsens with increasing time. The sclerotic bands are observed in the skin of the flexural areas of the axillae and antecubital and popliteal fossae after poikiodermatous changes which, in addition to marked poikiloderma, exhibits formation of extraordinary reticulated and linear, hyperkeratotic and sclerotic bands which extend across the flexures.
HSP may be inherited as autosomal dominant trait with incomplete penetrance and is most severe in males (1). In our patient, the family history showed that the patient's father and younger brother had poikilodermatous skin, but other members were not affected including grandfather and grandmother. The fact that his father is the first affected member of the family suggests that the disease in this male may represent a new mutation.
HSP may be also accompanied with cardiovascular abnormalities. Though cardiac involvement was reported in only two previously described patients (1, 3), several members of reported patient's family had cardiac valvular diseases or impending valvular diseases (1, 3, 4). Also, our patient's father died at 40 yr as a result of cardiovascular disease. These facts suggest that cardiac abnormalities may represent an important element in HSP. Therefore, it is necessary to evaluate regularly the existence of cardiovascular disease.
Clubbing is a physical sign characterized by bulbous enlargement of the ends of one or more fingers or toes. It usually acquired and is associated with cardiopulmonary and gastrointestinal diseases (5). Though it was observed in most HSP patients, no cardiopulmonary abnormalities were found to explain the clubbing in most previously reported patients except two above described patients who had cardiovascular diseases.
Other combined abnormalities included tissue calcinosis (1), micrognathia (2), maxillary bossing (2), mandibuloacral dysplasia (4), Raynaud's phenomenon (3) and growth retardation were also reported. Our case had micrognathia and growth retardation, but had no other above described diseases.
Recent article about HSP was reported in 2006 (6). It showed poikilodermatous change of skin associated with tendon contracture and progressive pulomonary fibrosis. In previous reports, although there were cardiac involvement, no pulmonary involvement was described. Also, her family had diffuse interstitial pulmonary fibrosis. The authors suggested that HSP might be devided into the three categories: Weary form HSP, HSP with cardiac involvement, and HSP with tendon/pulmonary involvement. However, further research is needed for that.
The histopathologic findings showed homogeneous & dense sclerotic collagen fibers in the upper dermis and fragmentations of damaged elastic fibers in the dermis. In our patient, increased melanin pigments in the basal layer and sclerotic changes and telangiectasia in the upper dermis were found in the H&E stain. Also, Elastic stain showed fragmentations of elastic fibers in the dermis. According to Greer's study (2), immunofluorescent studies revealed no evidence of immunoglobulin or complement deposition in the epidermis, basement membrane zone or other spaces in the dermis. Also, electron microscopic studies failed to demonstrate any specific abnormality of dermal connective tissues other than elastic fiber fragmentation.
The differential diagnosis of HSP consists of several diseases that are mandibuloacral dysplasia (4), hereditary acrokeratotic poikiloderma (7), poikiloderma congenital (8), xeroderma pigmentosa (9), dyskeratosis congenital (10), Werner's syndrome (11), and poikiloderma vasculare atrophicans. Poikiloderma without prior vesiculation or eczema distinguishes HSP from hereditary acrokeratotic poikiloderma, and autosomal dominant trait does HSP from poikiloderma congenital and xeroderma pigmentosa. HSP must be also distinguished from mandibuloacral dysplasia and Werner syndrome. Mandibuloacral dysplasia has poikilodermatous appearance and linear sclerotic bands, but it has mandibular hypoplasia, delayed cranial suture closure, dysplastic clavicles, and acroosteolysis (12). Although cutaneous manifestations associated with mandibuloacral dysplasia were similar to those associated with HSP, the extracutaneous manifestations in our patient were not found in association with mandibuloacral dysplasia. Also, unlike autosomal recessive inheritance visible from mandibuloacral dysplasia, pedigree of our patient showed autosomal dominant inheritance. Werner syndrome becomes apparent later, typically in the third or fourth decade.
To the best of our knowledge, 11 cases of HSP have been reported in the medical literature (1, 2, 3, 6, 13). Of these previously described patients, eight patients were African-Amerian (1, 2) and three were Caucasian (3, 6, 13). However, Korean patients have not been reported yet, therefore this is the first case of HSP occurred in Korea.


 XML Download
XML Download