

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pulmonary arterial hypertension (PAH), which is characterized by a progressive increase in pulmonary vascular resistance leading to right ventricular hypertrophy, right ventricular failure and death, is a devastating disease with a progressive course and poor prognosis (1, 2). In PAH, the median survival rate is considered to be 2.8 yr from the time of diagnosis (3). Although the pathogenesis of PAH is multifactorial, progressive endothelial cell dysfunction seems to play the most crucial role. This leads to an overproduction of vasoconstrictors and proliferative factors such as endothelin (ET)-1 and a concomitant reduction in vasodilators and antiproliferative factors such as prostacyclin and nitric oxide. Treatment strategies for PAH include long-term oxygen or inhaled nitric oxide, vasodilators, calcium channel blockers, intravenous prostacyclin, and ET receptor antagonists. However, all of these therapeutic options remain unsatisfactory as a result of high cost, limited effectiveness, or serious side effects. Therefore, a new treatment modality with long-term effects and less side effects is needed for the treatment of PAH.
RNA interference (RNAi) represents a recent breakthrough in effective blocking of the target genes in mammalian cells. Small interference RNA (siRNA) is oligonucleotides of around 21-23 nucleotide in length. Double-stranded RNA (dsRNA), small hairpin RNA (shRNA), and single siRNA can be specifically synthesized and delivered to the cell to target specific messenger RNA (mRNA). Vectors encoding shRNA with appropriate promoters are employed to generate siRNA within the cell to increase the efficacy of gene silencing. We used the lentiviral vector because this is known to induce prolonged RNAi with high transduction efficiency. siRNA is being explored as genetic inhibitors of gene expression, as well as potential therapeutics against inflammatory states, viral and hematological diseases, cancers and genetic disorders due to a dominant genetic effect, such as amyotrophic lateral sclerosis (4). Overall, these agents might be useful for a hypertensive state, but the efficacy of specific siRNA expression has not been tested in PAH (5).
ET-1 is a 21-amino acid peptide with potent vasoconstrictor and smooth muscle mitogenic properties. ET-1 is produced predominantly by endothelial cells but is also produced by leukocytes, macrophages, smooth muscle cells, cardiomyocytes and mesangial cells (6). It is produced from its inactive intermediate, big ET-1, through the action of endothelin-converting enzyme-1 (ECE-1). It is known to act through two types of receptors, ET receptor type A (ETA) and ET receptor type B (ETB). Plasma ET-1 levels are reportedly high in pulmonary hypertensive patients (7), and the ameliorative effects of ET receptor antagonists in a monocrotaline (MCT)-induced pulmonary hypertensive rats have been reported (8). These data suggest that endogenous ET-1 is involved in the progression of PAH and the blocking of ET function is expected to be effective in the treatment of this condition (9).
MCT is an alkaloid contained in the seeds of Crotalaria spectalalia, and its metabolite, MCT pyrrole, injures the endothelial cells of pulmonary blood vessels, causing severe pulmonary vascular disease in the absence of intrinsic heart and lung disease, thus suggesting its value as an animal model of idiopathic PAH (10). Despite its frequent use for many decades, the basic mechanism underlying PAH induction by MCT has not been fully understood.
In the present study, we investigated the therapeutic effects of shRNA targeting ECE-1 in MCT-induced pulmonary hypertensive rats by measuring not only pulmonary arterial pressure and right ventricular hypertrophy but also histopathological changes. Also, we performed a RT-PCR and western blot analysis in rat lung tissues in order to reveal the effectiveness of gene silencing.
MATERIALS AND METHODS
Ninty four pathogen-free male Sprague-Dawley (SD) rats (Orient Co., Seoul, Korea) weighing between 260 and 320 g were used in the present study. All animals were housed in climate-controlled conditions with a 12 hr light: 12 hr dark cycle, and had free access to chow and water.
The animal studies were performed after receiving approval of the institutional animal care and use committee (IACUC) in Ewha Womans University (IACUC approval No. 10-0141).
MCT (Sigma Chemical Co., St. Louis, MO, USA) was dissolved in 1 N HCl. The pH was neutralized with 0.5 N NaOH to pH 7.4 and the volume of the solution was adjusted with phosphate-buffered saline to achieve a concentration of 20 mg/mL. Pulmonary hypertensive rats were prepared by injecting the MCT at a subcutaneous dose of 60 mg/kg into the dorsum of the neck. Control rats were given by an equal volume of saline at the same site.
Because there have not been any reports investigating the effectiveness of target gene base sequence on ECE-1 mRNA (Genbank accession number NM_053596), we randomly selected five base sequences and investigated its effectiveness. The five base sequences are as follows.
We constructed shRNA using the above listed five base sequences and performed real time RT-PCR to investigate gene suppression effectiveness of ECE-1 on 9 L cell line and observed gene suppressions of 83%, 62%, 89%, 82%, 92%, respectively. We used HSP90AA as a nucleic acid and Luciferase as a control group to confirm ECE-1 expression. We ultimately selected the base sequence that showed 92% gene suppression as the target gene sequence of ECE-1 mRNA. The RNAi target sequence of ECE-1 mRNA (GenBank® accession number NM053596) was 5'-GACGCCATCTACAACATGATAGGCT-3'. Reduction efficiency of this sequence was confirmed to 92% by RT-PCR.
The manufacturing process entailed the following processes. The backbone transfer plasmid of the synthesized recombined lentivirus was combined with the core packaging plasmid and envelope plasmid onto 293T packing cell line (Invitrogen, Carlsbad, CA, USA) and transfected nucleic acid by calcium-phosphate co-precipitation. It was cultured for 72 hr and the supernatant was obtained after filtration with 0.45 µm membrane filter and immediately stored in -70℃. Viral titer was determined by infecting HeLa cells with lentivirus that expressed GFP and measuring the cells through FACS and measuring viral P24-gag protein using ELISA kit on the supernatant. We cultured 5 × 105 HeLa cells onto 6-cm dishes and took 1 mL of the culture medium that contained lentivirus that expressed GFP, prior to ultracentrifugation, and infected 5 × 105 HeLa cell line with DEAE dextran (10 µg/mL). We exchanged culture media after 16 hr and washed the cells 3 times with phosphate-buffered saline (PBS). After 48 hr, we treated the cells with trypsin and performed FACS analysis after fixating it for 30 min on 2% paraformaldehyde. We used HeLa cells that were not infected with the virus as the control group. We determined lentivirus titer by the number of HeLa cells that expressed GFP according to 1 ng of p24 protein.
Lentiviral vector containing shRNA targeting rat ECE-1 gene was constructed and propagated further in 293T cell. As a control vector, a scrambled shRNA (5'-AATCGCATAGCGTATGCCGTT-3'), which has no homology with mammalian mRNA sequences, was inserted in the lentiviral vector. Recombinant lentiviral GFP was tested for transduction efficiency, and 1.3 × 107/mL and 2.0 × 107/mL were calculated respectively under fluorescence microscope installing a digital camera system.
Experiment protocol
The ninty four 7-week-old SD rats were randomly divided as follows; the control group (n = 24) were injected subcutaneouly with normal saline, the MCT group (n = 35) with scrambled shRNA and MCT, and the shRNA group (n = 35) received intravenous ECE-shRNA (1 mL) through the rat tail vein. The MCT group and the shRNA group were subcutaneously injected with MCT 60 mg/kg into the dorsum of the neck. The number of the MCT group and the shRNA group rats is more than that of the control group because of early mortality (33%) during the previous study (11). On days 4, 7, 14, and 28 of the experiment, 6 rats in each group were anesthetized with Zoletil (Virbac, S.A, France) and Rompun (Bayer, Pittsburgh, PA, USA) intraperitonealy. After stable anesthesia, the right jugular vein and the right carotid artery were isolated, and polyethylene catheter (PE50) was inserted into the right ventricle (RV) and aorta. Mean right ventricular pressure (RVP) and systolic aortic pressure were measured. After the RV and aorta pressures were measured, blood samples were collected for circulating ET level determination. After exsanguination, the heart was excised. The atria were removed and the RV free wall was dissected from the left ventricle (LV) and interventricular septum (IVS) and the wet weights of the RV, LV including septum and lungs were measured. The RV-to-LV plus septum ratio (RV/LV + IVS) and RV-to-body weight ratio (RV/BW) in milligrams per gram were used as indices of right ventricular hypertrophy (RVH), while lung-to-BW ratio (lung/BW) in milligrams per gram was used as an index of pulmonary congestion. To perform a morphologic analysis of the lung, cannulas were inserted into the trachea and buffered formalin (10%) was infused for fixation of the lung. A part of lung tissues was cut into pieces, frozen in liquid nitrogen, and then were stored at -80℃ for future RT-PCR and western blot analysis.
Histopathological examination
To histologically investigate the lungs, the lungs were excised and immersed in formalin. Paraffin sections of 3 µm thickness were stained with hematoxylin and eosin and were examined under light microscopy. A computerized visual imaging system was used to photograph areas of interest, with the image digitized by using image-Pro plus 6.0 program. Measurements of medial thickness (MT) and vessel diameter of pulmonary arteriole were made digitally on 25-100 µm sized vessels in each photographic field by an observer blinded to experimental group. Measurements were made at random on 30 muscular arteries per lung sections. MT was calculated as MT (%) = medial wall thickness on either side/vessel diameter × 100. For the histological assessment of neomuscularization, we counted the numbers of the muscularized vessels with diameter < 50 µm in a × 200 magnification field below the terminal bronchiole. A total of 20 fields were examined in each rat for this analysis.
Serum endothelin-1 assays
Serum was collected to determine ET-1 concentrations. The serum ET-1 concentrations were measured using a human ET-1 immunoassay kit (QuantiGlo; R&D Systems, Minneapolis, MN, USA) by a sandwich enzyme immunoassay technique. A monoclonal antibody specific for ET-1 was pre-coated and immobilized onto a microplate, followed by samples with ET-1. After washing, an enzyme-linked monoclonal antibody specific for ET-1 was added to the wells. Following a wash to remove any unbound antibody-enzyme reagent, an enhanced luminol/peroxide substrate solution was added to the wells and light was produced in proportion to the amount of ET-1 bound in the initial step. A microplate luminometer was used to measure the intensity of the light emitted.
Real time reverse transcription-polymerase chain reaction analysis
Total RNA was extracted from the lung of rats. Total RNA (5 mg) was incubated with avian myeloblastosis virus reverse transcriptase in a mixture containing deoxynucleotides, random primer and RNase inhibitor at 42℃ for 60 min. cDNA was amplified in a final volume of 50 mL. The resulting first-strand of cDNA normalized by the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene. The normalized cDNA was used for the PCR procedure as a template. The specific primers for rat ET-1 were 5'-TCTCGGAGAGCAGAGACACA-3' (forward) and 5'-TGGACTTTGGAGTTTCT CCCT-3' (reverse). The specific primers for ETA were 5'-CACAGGCTTCAGTGTGC ATT-3' (forward) and 5'-CAACACAGGCCCTTAGC TTC-3'(reverse). The specific primers for ECE-1 were 5'-TCTCGGAGAGCAGAGACACA-3' (forward) and 5'-GTGCCACACCAAAACTACAG-3'(reverse). Amplification was conducted with a denaturing step at 94℃ for 30 sec; annealing at 58℃ for 30 sec and elongation at 72℃ for 30 sec for 36 cycles; and a final elongation step at 72℃ for 10 min. PCR products were seperated on a 10% (w/v) agarose gel and bands intensity were quantified with an image acquisition and analysis system (Bio-Rad, Hercules, CA, USA).
Western blot assay
Lung tissues were homogenized with extraction buffer (50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 5 mM dithiothreitol, 300 M phenylmethyl sulfonyl fluoride, 20 mM glycerophosphate, 1 mM NaF, 2 mM Na3VO4, 5 g/mL aprotinin, 5 M leupeptin, 1% Triton X-100, 10% glycerol, and 150 mM NaCl). Lysates were centrifuged (13,000 rpm, 10 min, at 4℃), and the supernatants collected were diluted 1:1 (v/v) with SDS sample buffer containing 40 mM Tris-HCl (pH 6.8), 8 mM EGTA, 4% 2-mercaptoethanol, 40% glycerol, 0.01% bromophenol blue, and 4% SDS, and then boiled for 7 min. Proteins were separated using 10% polyacrylamide SDS gels, and then transferred electrophoretically to a polyvinylidene fluoride membrane (Millipore, Bedford, MA, USA). The membrane was then blocked for 1 hr at room temperature with PBS containing 0.05% Tween 20 and 5% fat-free dried milk. The membranes were incubated with antibodies (ECE-1, ET-1, ETA, TNF-α, interleukine-6, caspase-3, Bcl-2, and VEGF) overnight at 4℃. Immune complexes were detected with horseradish peroxidase conjugated antibodies (Amersham-Pharmacia, Piscataway, NJ, USA) diluted 1:1,000 and incubated for 1 hr at room temperature. After application of the secondary antibody, blots were incubated in enhanced chemiluminescence kits (Amersham Pharmacia Biotechnology, Sanfrancisco, CA, USA) and exposed to photographic film.
Statistical analysis
All values were expressed as mean ± standard deviation. Differences in Kaplan-Meier survival curves between groups were evaluated by the log-rank test. Differences between all other parameters for the 3 groups were evaluated by ANOVA followed by multiple-group comparisons with the Bonferroni correction. A P value < 0.05 was considered statistically significant. Statistical analysis was performed using the Statistical Package for Social Science (SPSS 13.0) statistical software.
RESULTS
Effect of ECE-shRNA on survival
There were no deaths in the control group. There were 7 deaths in the MCT group, and 2 deaths in the shRNA group. Four-week survival was 59% in the MCT group and there was a significant increase in survival (88%) in the shRNA group (P = 0.012).
Gross findings
Gross appearance of the experimental rats was examined. There was no significant difference on gross appearance on days 4 and 7. The MCT group showed severe cardiomegaly, hepatomegaly, ascites and pleural effusion compared to the control group on days 14 and 28. The shRNA group showed less severe hepatomegaly, ascites and pleural effusion compared to the MCT group.
Hemodynamic parameters
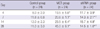
Compared to the control group, the MCT group showed a marked increase in mean RVP on days 4, 7, 14, and 28. Compared to the MCT group, the shRNA group showed a significant improvement in mean RVP on days 4, 7, 14, and 28 and especially showed a marked decrease of mean RVP by 68% on day 28 (14.5 ± 1.0 mmHg vs 45.0 ± 9.1 mmHg, P = 0.001) (Table 1).
There was no significant difference in mean arterial pressure (data was not shown).
Organ weight
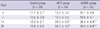
Body weight in the MCT group was lower than in the control group on days 4, 7, and 28. MCT group showed an increase in RV/(IVS + LV) values on days 14 and 28 compared with control group (Table 2), indicating marked RVH. A marked increase was observed in lung weight on days 4, 14, and 28 in the MCT group (Table 2), demonstrating the development of inflammatory injury in the lungs and the presence of pulmonary congestion.
Lung/body weight was significantly increased in the MCT group compared with the control group. There was no significant difference in the shRNA group compared with the MCT group (Table 2).
Histopathological examination
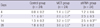
The MCT group showed a significant increase in medial wall thickness on days 14 and 28. The shRNA group showed significant reductions in medial thickness of vessels 25-100 mm on days 14 (26.3 ± 6.9% vs 29.3 ± 8.9%, P = 0.045) and 28 (28.5 ± 6.8% vs 39.0 ± 4.3%, P < 0.001) compared to the MCT group, but the values were still significantly greater than corresponding values in the control group (Table 3, Fig. 1).
The number of muscular intra-acinar arteries in the MCT group was significantly increased compared to the control group on days 4, 7, 14, and 28 and significantly decreased in the shRNA group compared with the MCT group on day 14 (2.5 ± 0.3 vs 3.2 ± 0.5, P = 0.048, Table 4).
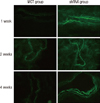
The control group without lentiviral transduction showed no fluorescence, whereas groups transduced with lentivirus encoding scrambled shRNA or shRNA targeting ECE-1 showed uniform endothelial expression of GFP, demonstrating a lentiviral transduction was efficient and well preserved throughout the whole experiment (Fig. 2).
Plasma ET-1 level
Four weeks after MCT injection, the mean plasma ET-1 concentration in the MCT group was significantly increased as compared to the control group (3.02 ± 0.99 pg/mL vs 1.34 ± 0.39 pg/mL, P = 0.004). In the shRNA group, the plasma ET-1 levels were lower than those in the MCT group, but did not reach significance.
RT-PCR analysis
The mRNA expression of ECE-1, ET-1, and ETA in the lung tissues were significantly increased in the MCT group in weeks 2 and 4. The mRNA expression of ET-1 and ETA was significantly decreased in the shRNA group in week 4 compared to the MCT group (ET-1, 0.76 ± 0.07 vs 0.85 ± 0.05; ETA, 0.82 ± 0.07 vs 0.94 ± 0.06, P = 0.011), and other gene expressions in the shRNA group were lower than those in the MCT group, but did not reach significance (Fig. 3).
Western blot analysis
The protein levels of ECE-1, ET-1, ETA, TNF-α, interleukin (IL)-6, caspase-3, Bcl-2, and VEGF in the lung tissues were significantly increased in the MCT group in weeks 2 and 4. The protein levels of ETA in the lung tissues were significantly decreased in the shRNA group in week 2 compared to the MCT group (0.79 ± 0.04 vs 0.90 ± 0.15, P = 0.012) and ECE-1, ET-1 protein levels in shRNA group were lower than those in the MCT group, but did not reach significance (Fig. 4). The protein levels of TNF-α (P < 0.001) and VEGF (P < 0.001) were significantly decreased in the shRNA group compared with the MCT group in week 4 (Fig. 5).
DISCUSSION
The present study showed that recombinant lentiviral vectors carrying shRNA targeting ECE-1 in MCT-induced pulmonary hypertensive rats could effectively reduce the mean pulmonary arterial pressure, pulmonary vascular remodelling and mortalities. As far as we are aware, this is the first report to demonstrate the effectiveness of RNAi method in the treatment of MCT-induced pulmonary hypertensive rats.
Animal models of PAH have contributed to our understanding of the underlying mechanism and have aided in the development of therapeutic targets. The most widely used models are the chronic hypoxia and the MCT-induced rat models of PAH (12). In the present study, we first measured the hemodynamic parameters of MCT-induced pulmonary hypertensive rats by a cannula method. The systolic RVP in the MCT rats increased with time after MCT injection. Determination of weights revealed greater increase in the RV/(LV + IVS) ratio and lung weight on days 14 and 28 after MCT administration. Since the ratio of RV/(LV + IVS) is considered to be a marker of RVH, augmentation of this ratio may represent the genesis of RVH and PAH. Accordingly, these findings confirmed the MCT was an appropriate tool for the induction of PAH. The MCT model was introduced 50 yr ago (13). It is based upon a single injection of MCT intraperitonealy or subcutaneously which rapidly leads to severe pulmonary vascular disease in the absence of intrinsic heart and lung disease, thus suggesting its value as an animal model of idiopathic PAH (14). Despite its frequent use for many decades, the basic mechanism underlying PAH induction by MCT remains poorly understood. Hill et al. (15) reported that the pathogenesis of MCT-induced PAH is multifactorial, consisting of vasoreactive, inflammatory, cytokine and hypoxic components. Miyauchi et al. (16) reported a single injection of MCT caused a progressive increase in systolic RVP that was significantly higher than the control value by day 14, after which it rose quite rapidly. However, our results showed mean RVP was increased on day 4 and rose rapidly by day 28. Major differences exist between rat strains with regard to their MCT sensitivity and even within a given strain the inter-individual differences, its time of onset and extent of toxic effects can vary markedly (10). These differences in susceptibility may relate to the pharmacokinetics of MCT, and possibly include differences in degradation and hepatic formation of the toxic MCT pyrrole or conjugation and excretion (14).
Yanagisawa et al. (17) discovered ET-1 more than 20 yr ago and it was revealed that its release could have a major role in the pathogenesis of a variety of cardiovascular diseases including PAH. ET-1 could contribute to the development of human PAH through its strong vasoconstrictive and promitogenic properties. Increased plasma levels of ET-1 were reported in experimental models of PAH and some patients with PAH (18). The ET system can be antagonized by inhibition of the ECE-1 and by the blockade of ET receptors. Pharmacological blockade of ET synthesis, by inhibition of ECE-1, is an intuitive approach in reducing the elevated activity of ET-1 associated with numerous cardiovascular disease states. The viability of this strategy was demonstrated by the ability of phosphoramidon to cause vasodilation and attenuate big ET-1 induced vasoconstriction in healthy volunteers (19). Since then, a variety of animal studies and preliminary work in human patients have shown potentially beneficial effects of ECE inhibition (20). However, ECE inhibitors have been eclipsed by ET receptor antagonists in both experimental and clinical use. The limited enthusiasm for ECE inhibition can be explained by theoretical concerns, which is the redundancy in ET-1 generating pathways and the presence of non-ECE big ET-1 processing enzymes. However, Takahashi et al. (21) reported that ECE inhibitor protected the development of RV overload and medial thickening of pulmonary arteries nearly equivalently to that of ETA receptor antagonist in rats with MCT-induced PAH. However, nearly all drugs for PAH have a short half-life and some kinds of side effects. Therefore, a new therapy with long-term effects and less side effects is needed for a disease requiring treatment.
RNAi is a gene silencing phenomenon discovered in the last 10 yr (22). This discovery creates a new gene down-regulation technique. RNAi, triggered by double strand RNA, features high specificity, only degrading mRNA with sequence homology. Vector-mediated shRNA can induce sustained RNAi in comparison to siRNA. Lentiviral vector is known to induce prolonged RNAi with high transduction efficiency (23). Lentiviral delivery of siRNA represents a powerful tool for functional genomics. The advantages of lentivirus for gene transfer, especially to non-dividing cells, make the RNAi technique more accessible for specific in vivo silencing in mammalian cells (5). Effective methods for the delivery of siRNA/shRNA to allow a sufficient silencing effect in the target organ are yet to be developed (24).
"High-pressure injection" was the first strategy to demonstrate successful delivery of siRNA in vivo. A large volume (1-2 mL) of saline containing unmodified siRNA is injected intravenously into the tail vein within a short time, which presumably results in the siRNA molecules being forced into several organs. In the present study, we injected the constructed and propagated lentivirus (1 mL) containing shRNA targeting rat ECE-1 gene through tail vein and revealed that this method effectively attenuated the PAH and vascular remodeling in rats with MCT-induced PAH when compared to other studies (25-27). As far as we are aware, this is the first report to demonstrate the effectiveness of RNAi targeting ECE-1 gene using lentivirus vector.
A variety of methods have been used to assess the different aspects of pulmonary vascular remodeling. Medial wall thickening of pulmonary arteries and neo-muscularization of normally non-muscular arteries can be assessed histologically in lung section stained for smooth muscle and elastin or immunolabelled with antibodies of smooth muscle alpha-actin (28). Medial thickness of arteries in the histological sections is quantified by measuring wall thickness, as delineated by the internal and external elastic lamina and expressing it as a percentage of vessel diameter (29). The histological assessment of neomuscularization involves counting all of the vessels with diameters < 50 mm in a complete lung section and determining the percentages of vessels that are fully muscularized, partially muscularized, and non-muscularized (30). Increased thickness of the pulmonary arterial media is believed to play a major role in the development of MCT-induced PAH and sustained irreversible increases in pulmonary vascular resistance. ET-1 has potent proliferative activity on vascular smooth muscle cells. In the present study, decreased ET-1 levels and improved histological examination revealed that lentiviral vector-mediated shRNA targeting ECE-1 effectively prevented arterial medial thickening in the MCT rats, suggesting that ET-1 participates in the thickening of pulmonary vessels. Therefore, it is conceivable that the inhibition of PAH by shRNA targeting ECE-1 is, at least in part, due to the antagonism of ET-1 induced vascular proliferation. RVH occurred with the development of PAH after MCT treatment. Since ET-1 was shown to induce myocardial cell hypertrophy, the increase in ET-1 production in the heart may also contribute to an exaggeration of cardiac hypertrophy after day 14. Increased ET-1 protein contents in the heart and lung tissues were noted on days 14 and 28.
Additionally, measurements of pulmonary vessel wall thickness and the number of muscularized small intrapulmonary arteries revealed that the pulmonary vascular remodeling was improved in rats treated specific shRNA. The therapeutic efficacy of specific shRNA may thus be attributable to its inhibition of ET-1 induced pulmonary vascular smooth muscle cell proliferation. Nevertheless, this specific shRNA did not completely ameliorate the pulmonary vascular remodeling in MCT treated rats. This fact implied that other mediators may also be involved in MCT-induced pulmonary vascular remodeling, such as vascular endothelial growth factor, inflammatory cytokine and angiotensin II.
A RT-PCR and western blot analysis in rat lung tissue were performed in order to reveal the effectiveness of gene silencing. MCT increased the mRNA expression and the protein level of ECE-1, ET-1, and ETA of the lung in weeks 2 and 4, and lentiviral vector-mediated shRNA targeting ECE-1 significantly decreased the mRAN expression of ET-1 and ETA in week 4 and the protein level of ETA in week 2. ECE-1 gene expressions and protein levels were lower than those in the MCT group, but they were not significant. Considering the fact that the mortality, hemodynamic and histopathologic results are improved, this is somewhat unexpected. Although RNAi is highly specific in knocking down the expression of genes, there are considerable issues rising in regards to off-target effects (24). The most common intrinsic off-target effect induced by siRNA is caused by the failure to identify similar sequences with only few difference in other genes that induce unspecific silencing. Another common intrinsic off-target effect provoked by siRNA is the activation of intracellular protein kinase and immune pathways that are linked to Toll-like receptor activation (31).
We performed TNF-α, IL-6, caspase-3, Bcl-2, and VEGF protein including ECE-1, ET-1, and ETA by western blot analysis to investigate the off-target effect of shRNA targeting ECE-1 injection. We demonstrated that there was decreased TNF-α and VEGF gene expressions after the shRNA injection in week 4. These suggest off-target effect of shRNA targeting ECE-1 injection.
High pressure injection can cause significant damage to the integrity of the normal tissues. Although RNAi has tremendous potential, it also possesses several intrinsic risks that could be dangerous and thus, the importance of carefully designing shRNA constructs to optimize the dose and the exact sequence of siRNA expression cannot be overemphasized (32). These emerging methods are still under investigation and need to be thoroughly investigated before being used in therapeutic applications.
In summary, we demonstrated that recombinant lentiviral vectors carrying shRNA targeting ECE-1 prevented PAH and RVH and exerted an anti-proliferative effect on pulmonary artery smooth muscle cells. Although the precise mechanism of the dramatic inhibitory effects is unclear, these findings suggest the possibility that this treatment modality could be effective against hemodynamic, histopathological and gene expressions changes in PAH.





 XML Download
XML Download