

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Happle et al. (1) first proposed the term phacomatosis pigmentokeratotica (PPK), which is characterized by organoid nevus with sebaceous differentiation arranged along Blaschko's lines, and speckled lentiginous nevus (SLN) arranged in a checkerboard pattern. Happle (2) defined that this syndrome represented an example of the genetic mechanism didymosis, or twin spotting.
PPK is divided into two subgroups according to the presence or absence of extracutaneous involvements. It is very rare that approximately 30 cases of PPK have been reported (3). In the Korean literature, there are no previously reported cases of PPK without extracutaneous manifestation. Herein, we report a rare case of PPK in a preterm baby.
CASE DESCRIPTION
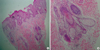
On November 10, 2010, the patient in our study was born at 27 weeks' gestation after a pregnancy complicated by a premature rupture of the membranes and weighed 820 g. At birth he was reported to have yellow-pink grouped papules and plaques involving the face, scalp and right side of the trunk. Upon examination, the skin abnormalities were diagnosed as extensive epidermal nevus. The epidermal nevus had sebaceous features and the configuration was in accordance with Blaschko's lines (Fig. 1A, B). At seven days of age, the erythema appeared on the right abdomen and arm which gradually darkened and became slightly raised similar to a warty lesion. By the age of one year, multiple tiny raised pigmented lesions also appeared on the upper back and chest. The pigmented lesions on the upper back and chest had gradually extended. The appearance was similar to a large melanocytic nevi in a speckled lentiginous pattern (Fig. 1C, D). Histological examination of a skin biopsy taken from the right side of the cheek showed compact hyperkeratosis, moderate papillomatosis and acanthosis. There was a substantial increase in the number of mature sebaceous lobules, hair follicles and eccrine glands in the epidermis. It was compatible with an organoid nevus with sebaceous differentiation (Fig. 2). Correlating the observed clinical presentation with the histopathological findings, the diagnosis of PPK was established. The infant has neither remarkable echogenic lesions in the caudothalamic groove nor asymmetric ventriculomegaly or IVH on the brain sonography at one month after birth. Systemic examination, laboratory findings, bone x-ray, and karyotypical analysis revealed no abnormalities. In addition, the family history was noncontributory. At the one year follow-up examination, he was healthy and had no impaired development of the neurological system. Treatment with topical budesonide improved the inflammation of the erythematous plaques; however, the plaques themselves showed no change.
DISCUSSION
PPK, as first described by Happle et al. (1), refers to a clinical syndrome consisting of SLN, organoid nevus with sebaceous differentiation and certain neurologic, ophthalmologic, and skeletal anomalies. This appearance can best be explained as the co-occurrence of the two different nevi reflecting a twin spot phenomenon (2). The two components of this twin spot phenomenon have been identified as the Schimmelpennning syndrome and SLN syndrome (3). The Schimmelpenning syndrome is defined as the association of the linear nevus sebaceous arranged along the Blaschko's lines and extracutaneous involvement, including skeletal defects, neurologic defects such as hemimegalencephaly with contralateral motor disease, and ocular abnormalities, including coloboma and epibulbar lipodermoid (4). PPK shares the epidermal nevus with sebaceous differentiation with the Schimmelpenning syndrome, but it differs from the latter in the presence of SLN and the absence of major central nervous system effects, including conjunctival lipodermoid, or coloboma.
Approximately 30 cases of PPK have been reported (3). PPK is commonly considered to be associated with extracutaneous abnormalities such as neurologic (hemiatrophy with variable muscle weakness, dysesthesia, and hyperhidrosis in the region of the SLN), ophthalmologic (sclerotic nevus spread, internal strabismus, and ptosis) and skeletal disorders (postural deviation with the appearance of kyphosis/scoliosis) (5). However, unlike other reported cases, our case had no extracutaneous abnormalities.
In conclusion, our patient represents a rare case of PPK without systemic involvement. To our knowledge, this case is one of only nine cases reported in the literature. Eight of all reported cases have no extracutaneous abnormalities described (6-9). Therefore, PPK could be divided into two subgroups by the presence or absence of systemic involvement. Also, some cases of PPK with no extracutaneous abnormalities developed basal cell carcinoma in the nevus sebaceous (9, 10). Therefore, physicians should not neglect to maintain adequate patient follow-up care to ensure early detection of any possible associated malignant change.

 XML Download
XML Download