

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Any genomic imbalances, deletions or duplications of an X chromosome have a much more severe impact on males than females. With this in mind, idic(Xp), i(Xq), r(X) are only observed in live-born females. In males, most cytogenetically visible deletions of the X chromosome involve the terminal portion of Xp (Xp22.2→Xpter), which leads to nullisomy of the deleted region, has been recognized as a cause of variable contiguous gene syndromes (1, 2). The phenotypes depend on the extent and position of the deletion, showing the variable association of apparently unrelated clinical manifestations such as Leri-Weill dyschondrosteosis (SHOX), chondrodysplasia punctata (CDPX1), mental retardation (NLGN4), ichthyosis (STS), Kallmann syndrome (KAL1), ocular albinism (GPR143). The extent of terminal Xp deletions is limited by the presence of male lethal genes in Xp22.2 at about 10-11 Mb from the telomere. The majority of the deletions in viable reported males extended to the STS (-7.0 Mb) or to the KAL1 (-8.5 Mb) locus. Larger Xp deletions extending beyond the KAL1 locus are very rare (3). To the best of our knowledge, there are only two documented reports which are the most severe known male terminal Xp deletions extending to the OA1 locus (3, 4). Females with similar Xp22 deletions are phenotypically normal except for short stature, because they need only one active copy of this region to be normal. Herein, we present the clinical, cytogenetic, and microarray study of a boy and sister with deletions of Xp (Xp22.2→Xpter). As far as we know, this is the first reported case of a male with a 9.7 Mb terminal Xp deletion including the OA1 locus in Korea.
CASE DESCRIPTION
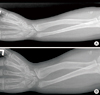
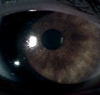
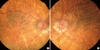
A 13.5 yr old boy was visited for evaluation of short stature, on 9 April 2007. He was born at term with weighing 3,500 g by uncomplicated spontaneous vaginal delivery, the first child of unrelated parents. At 5 yr old, he had undergone surgery due to a cleft lip. In addition, delayed motor and language development was observed. With regards to his physical stature, height was 124.8 cm (< 3 rd percentile), Upper segment/lower segment ratio 1.3 (normal 1.0), and Weight 43 kg (50th percentile). Furthermore, body mass index (BMI) was 27.6 kg/m2 (95-97th percentile). Both rhizomelic and mesomelic limb shortness and terminal phalanges of fingers shortness were present. He had a flat and round face with midface hypoplasia, a broad flat nose, a thin upper lip with post operation scar. Brown scales covered the extremities, trunk, neck, preauricular areas (Fig. 1). He had not yet attained puberty as he exhibited; absence of pubic hair, small penis and scrotal sac. Laboratory findings were shown as follows: hemoglobin 13.8 g/dL, white blood cell count 7,900/µL, platelet 338,000/µL, aspartate transaminase (AST) 61 IU/L, alanine transaminase (ALT) 85 IU/L, total cholesterol 271 mg/dL, triglyceride 244 mg/dL, low density lipoprotein cholesterol (LDL-C) 200.7 mg/dL, high density lipoprotein cholesterol (HDL-C) 53.7 mg/dL. There was no elevation in Luteinizing hormone (0.1 mIU/mL), Follicle stimulating hormone (0.46 mIU/mL) and Testosterone (0.1 ng/mL). Prepubertal response was observed in a Gonadotropin-releasing hormone (GnRH) stimulation test. Radiographs showed shortness of the long bones, bowed radii, and Madelung deformity of forearms (Fig. 3A). Bone age (Greulich-Pyle method) was 13-14 yr at the chronological age of 13.5 yr. Brain MRI revealed almost hypoplastic olfactory bulb with an ill-defined olfactory tract and sulci. Abdominal US revealed a fatty liver. He had hyposmia which was tested by using serial dilutions of multiple odorants. He had impaired vision with nystagmus. Ophthalmological examination disclosed iris transillumination demonstrating defects in the iris pigment epithelium (Fig. 4), hypopigmentation of the fundus, and macular hypoplasia in both eyes (Fig. 5). Wearing tinted glasses with correction of myopic astigmatism and a hat was recommended in order to reduce the risk of exposure to sunlight. Psychometric evaluation showed a total IQ of 52 (WISC-R) indicating mild mental retardation. Also, he was impulsive, exhibited hyperactive behavior including poor attention. Cytogenetic analysis revealed a terminal deletion of Xp; 46,Y,del(X)(p22.2). To specify the breakpoint, high resolution microarray analysis performed. We analyzed the genomic DNA on the Affymetrix Cytogenetics Whole-Genome 2.7M array, which provides whole genome coverage with a high density of 2.7 million markers. This array contains approximately 400,000 SNP markers and 2.3 million non-polymorphic markers, with high density coverage across cytogenetically significant regions. Genomic DNA was extracted from peripheral blood using QIAamp DNA mini kit DNA Isolation Kits (Qiagen, Hilden, Germany). The microarray assay was performed according to the manufacturer's protocols (http://www.affymetrix.com) (Affymetrix Inc., Santa Clara, CA). Data was collected using GeneChipÒ Scanner 3000 and CEL Files were analyzed using Affymetrix Chromosome analysis Suite software (ChAS v.1.1). The results revealed a terminal 9.7 Mb deletion at Xp22.2 resulting in the absence of genes from the telomere of Xp to GPR143 of Xp22 (Fig. 6).
At 14.5 yr, his height was 125.7 cm. This showed a gain of 2 cm for 1 yr. But the growth hormone provocation test showed no deficiency of growth hormone. In addition, he did not show any pubertal change, thus we began a monthy injection of testosterone enanthate at a dosage of 50 mg increasing the dosage by 50% every 6 months. At 16.3 yr, his height was 134.4 cm. This demonstrated a gain of 8.7 cm for 1.75 yr. However, he didn't take any medications nor follow any recommended diet for his fatty liver and hyperlipidemia, and he also stopped visiting our clinic. At 18 yr, he visited again. His height was 135.5 cm (< 3 rd percentile), his weight was 61 kg (25-50th percentile), and his body mass index (BMI) was 33.2 kg/m2 (> 97th percentile). Laboratory findings were shown as follows: Fasting glucose 118 mg/dL, HbA1C 7%, AST 165 IU/L, ALT 152 IU/L, total cholesterol 287 mg/dL, triglyceride 560 mg/dL, LDL-cholesterol 189.8 mg/dL, HDL-cholesterol 40.0 mg/dL, Luteinizing hormone 0.17 mIU/mL, Follicle stimulating hormone 0.15 mIU/mL, Testosterone 0.2 ng/mL. In addition to his previous diseases, he was also diagnosed with diabetes mellitus.
Next, his sister was 11 yr old. Her personal physical characteristics are as follows: Height was 126 cm (3rd percentile), Weight 40 kg (70th percentile), Body mass index (BMI) 25.2 kg/m2 (>97th percentile). Both rhizomelic and mesomelic limb shortness were present (Fig. 2). She showed breast development, but no icthyosis. She had normal vision with myopia, showed slightly hypopigmented retina. Laboratory tests revealed hypercholesterolemia (total cholesterol 232 mg/dL), the remaining serum electrolytes and complete blood cell counts were normal. Radiological examination of the forearms and wrists confirmed Madelung deformity with lateral and dorsal bowing of the radius and ulna, distal ulna dislocation, and shortness of the long bones (Fig. 3B). Bone age was 11-12 yr at the chronological age of 11 yr. Cytogenetic analysis revealed a 46,X,del(X)(p22.2). Microarry analysis revealed same with the brother's. We bgan a growth hormone therapy (Eutropin 1.0 IU/kg/week). At 12.5 yr old, Menarche was observed. At 14 yr old, her final height was 143 cm. We couldn't confirm parent's cytogenetic analysis, because they were divorced. According to relatives, her mother was short. The mother's clinical picture closely resembled that of her sister. It suggests these patients inherited this aberrant deleted X chromosome from their mother.
DISCUSSION
Our patient's Xp deletion approximates the largest of Xp22 to ever have been reported in a male. He had distinct abnormalities that can be explained by this deletion. His phenotype represented a contiguous gene syndrome comprising clear clinical signs of Leri-Weill dyschondrosteosis (SHOX), chondrodysplasia punctata (CDPX1), mental retardation (NLGN4), ichthyosis (STS), Kallmann syndrome (KAL1), ocular albinism (GPR143). On the other hand, his sister's phenotype represented only Leri-Weill dyschondrosteosis. She did not manifest other symptoms, presumably due to preferential X chromosome inactivation of the structurally abnormal X chromosome. Due to this preferential inactivation, such X chromosome abnormalities have less of an impact on phenotype. As a result of inactivation, most genes on one X chromosome are silenced. However, some genes escape inactivation and were expressed from the active and inactive X chromosome. Genes in the pseudoautosomal regions (PAR) on both ends of the X and the Y chromosomes were not subject to X-inactivation. and many more genes escape inactivation on the distal Xp rather than Xq. Dosage imbalance of the genes in the PAR regions and genes that escape X inactivation contributed to the phenotypes in females with an X chromosome anomaly (5, 6). SHOX (Short Stature Homeobox-containing gene) was located in the PAR1 on the distal end of the X and Y chromosome at Xp22.3 and Yp11.3. Since genes in PAR1 do not undergo X inactivation, healthy individuals expressed two copies of the SHOX gene. SHOX haploinsufficiency were found in patients with idiopathic growth retardation and syndromic short stature (e.g. Turner syndrome, Leri-Weill dyschondrosteosis, Langer mesomelic dysplasia (7-9). Leri-Weill dyschondrosteosis (OMIM 127300) affects both sexes. Pubertal development and fertility are generally normal. Clinical characteristics include short stature, mesomelic shortening of the forearms and lower legs, and Madelung deformity (shortening and bowing of the radius with dorsal subluxation of the distal ulna) of the wrists. The growth-promoting effect of Growth Hormone therapy seemed to be equal to the effect seen in Turner syndrome (10). X-linked recessive chondrodysplasia punctata (CDPX1, OMIM 302950), due to mutations of arylsulfatase E (ARSE) gene, was a congenital disorder characterized by abnormalities in cartilage and bone development (11). It was characterized by the presence of stippled epiphyses on radiograms in infancy and early childhood, hypoplasia of the midface and of nasal bone, short stature, brachytelephalangy, and ectopic calcifications. The hypoplasia of distal phalanges of the fingers is a valuable aid in the diagnosis at an age when epiphyseal stippling is no longer present (12). In a number of patients with Xp22.3 contiguous gene syndromes the presence of mental retardation was reported and a locus for X-linked non-specific mental retardation (MRX) was therefore tentatively assigned to Xp22.3. NLGN4 (neuroligin 4, OMIN 300495) has been implicated in X-linked autism and mental retardation (13, 14). X-linked recessive ichthyosis (XLI, OMIM 308100) is an inherited disorder of keratinization due to steroid sulfatase (STS) deficiency (15). Accumulation of cholesterol sulfate in the epidermis leads to barrier instability and inhibits the desmosomal degradation, which is required for normal desquamation. Tightly adherent, brown, polygonal scales covered the extensor surfaces of the extremities, trunk, neck, scalp, and preauricular areas. KAL1 gene was defective in patients with Kallmann syndrome (OMIM 308700), characterized by isolated hypogonadotropic hypogonadism associated with anosmia or hyposmia (16). OA1 gene (GPR143) has been recognized as the gene underlying X-linked ocular albinism (OA1; OMIM 300500), which was mainly effective in pigment production within the eye, resulting in hypopigmentation of the retina, nystagmus, strabismus, foveal hypoplasia, abnormal crossing of the optic fibers, and reduced visual acuity (17). In addition to the genes discussed above, some patients have been reported to have symptoms regarding poor attention, hyperacivity, and impulsivity (18, 19). The region of Xp22.2-Xpter, excluding the pseudoautosomal region, contains other known or putative genes for which the clinical consequence of nullisomy is not yet known. Thus, further molecular and clinical studies of patients with Xp22.2 contiguous gene syndromes are needed.



 XML Download
XML Download