

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Myotonic dystrophy (DM) is a multisystemic disease whose most prominent manifestations are myotonia, atrophy, and progressive weakness of neuromuscular systems. DM1 is the most common adult myopathy. The prevalence are different in different geography, but it ranges from 1:100,000 to 1:10,000 (1). Although there is no information on the prevalence of DM1 in Korea, many cases have been reported (2-4). DM1 is caused by an expansion of an unstable CTG trinucleotide repeat in the 3° untranslated region (UTR) of the gene DMPK (myotonic dystrophy protein kinase) (5, 6). The gene is located on chromosome 19q13.3. Normal individuals have between 5 and 37 CTG repeats (3). DMPK alleles greater than 37 CTG repeats are unstable and may expand in length during meiosis and mitosis. This results in increasing disease severity and decreasing age of onset in successive generations (7, 8). Most of the congenital DM1 patients had more than 1000 CTG repeats and showed severe generalized weakness, hypotonia and respiratory compromise after immediate birth.
There has been no reported case of congenital DM1 with premature birth in the 30th week of gestation.
CASE DESCRIPTION
This case presented 2 newborn nonidentical twins from unrelated parents. The pregnancy was achieved by in vitro fertilization (IVF) due to sterility of the mother. The mother was 31 yr old, and gravida 3 para 0 living 0. She sometimes felt fatigue but she was healthy. At the prenatal ultrasound examination, bilateral lateral ventricle dilatation from the first fetus and polyhydramnios and prominent stomach from the second fetus were detected. There was a preterm labor in the 30th week of gestation and the twins were delivered with the emergency Caesarian section due to fetal distress.
The first twin was born at 30+4 weeks of gestation on November 21, 2009. The body weight was 1.46 kg and the Apgar score was 1, 2, and 4 at 1, 5, and 10 min. At birth, he showed no respiratory effort, no movement, bradycardia and generalized cyanosis, so he was resuscitated with intubation and intratracheal epinephrine. He had a generalized edema. The face was expressionless and triangular with low-set ears and an inverted v-shaped upper lip. There was no spontaneous respiratory effort. He had distended and tense abdomen with decreased bowel sound. Single umbilical artery was noted. Severe hypotonia was significant. Primitive reflexes were not induced. Bilateral cryptorchidism was present. He was extubated on day 4, but reintubated due to multiple episodes of apnea with collapsed lung. He was maintained with mechanical ventilator until day 89, on which he could be weaned to nasal CPAP.
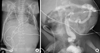
A chest X-ray on day 1 of the first twin showed bilateral pleural effusion and the amount has increased progressively over time (Fig. 1A). The pleural fluid analysis showed a white blood cell count of 2,480/µL with 94% lymphocytes. It also showed the fluid protein level of 2.7 g/dL, glucose level of 206 mg/dL, cholesterol level of 60 mg/dL, and a triglyceride value of 495 mg/dL, which confirmed the chylothorax. It was managed by chest tube drainage, octreotide, and medium chain triglyceride milk. Chest tube was removed at day 48. Hyperglycemia to 300 mg/dL was controlled by insulin. Creatine kinase level was 281 U/L.
PCR-Southern DNA analyses by biotin-(CTG)10 probe, using peripheral blood lymphocytes from the twins and both parents, resulted in the diagnosis of congenital DM1 in the twins. Father had both normal alleles with a CTG repeat length of 13 and 23. However, mother showed a CTG repeat length of about 150 and the twins showed a CTG repeat length of more than 750.
Orogastric tube feeding was started on day 4, but he was complicated by meconium ileus and necrotizing enterocolitis. Although the orogastric feeding was restarted on day 25, relative gut atony led to difficulties in establishing enteral feeds (Fig. 1A). However, this was eventually tolerated by day 40 using metoclopramide. Oral feeds were impossible with problems in which there was no effort to suck or swallow.
Brain ultrasound showed intraventricular hemorrhage grade II-III and periventricular leukomalacia. Intraventricular hemorrhage was complicated by hydrocephalus. Since neurosurgical intervention could not be done, intermittent spinal tapping was practiced to decompress cerebral pressure. Anticonvulsants were given to control generalized clonic seizure.
Although the first twin was tolerated by oxygen therapy after day 95, he was complicated by respiratory failure with pneumonia and expired on day 101.
The second twin weighted 1.33 kg and the Apgar score was 3 and 5 at 1 and 5 min. The second twin had milder symptoms and clinical outcomes than the first twin. At birth, he showed initial crying, though weak, but his respiratory effort was undetectable after a few seconds. Resuscitation and mechanical ventilation was started. There were clinical features strongly suggestive of congenital DM1; inverted V-shaped upper lip, poor respiratory effort, severe hypotonia, and paucity of spontaneous movements. Both arms and legs were thin and long. Bilateral cryptorchidism was noted.
On day 1, respiratory distress syndrome was confirmed and surfactant was given. Extubation was failed because of severe apnea on day 4 and intubation was maintained until day 38. Nasal CPAP was maintained by day 55. He showed improvement in respiratory function slowly. The second twin showed milder hypotonia than the first twin and faster improvement in spontaneous movement. Creatine kinase level was 194 U/L.
Orogastric tube feeding was started on day 4, but was intolerable due to vomiting and abdominal distension. After gastrograffin enema on day 21, all gastric symptoms regressed and spontaneous defecation was possible (Fig. 1B). After 38 weeks of corrected age, his sucking and swallowing efforts were improved. He could feed completely orally.
He showed intraventricular hemorrhage grade II-III complicated by hydrocephalus on day 7. Intermittent spinal tapping was attempted and anticonvulsants were given. On day 102, 44 weeks and 7 days of corrected age, he could be discharged home. Unfortunately, his parents quitted outpatient clinic visitation and it was reported that he was expired at home on day 215 with an apparent respiratory problem.
The twins' mother was healthy. However, after delivery, she was complicated by uterine atony, increased fatigue, hypomobility, retinal abnormality and cardiac arrhythmia.
DISCUSSION
For diagnosis of DM1, PCR analysis is used to detect repeat lengths less than 100 and Southern blot analysis to detect larger expansions. In our case, the 2 twins and their mother was diagnosed by Southern blot analysis. The mother who presented much milder symptoms than her babies had 150 repeat lengths. On the other hand, the twins showed more than 750 repeat lengths. In this case, polyhydramnios before birth and severe generalized weakness, hypotonia, and respiratory compromise after delivery were main features. They also had an inverted V-shaped upper lip and facial weakness with poor sucking.
For DM1, mortality from respiratory failure is high. Campbell et al. (9) described that a 25% mortality rate had been noted in the children with > 30 days ventilation, whereas no child in < 30 days ventilation died. Failure to thrive, club feet, and feeding difficulties are common problems, but surviving infants experience gradual improvement in motor function, swallowing, and respiration. In our case, the second twin reached full oral feeding with good swallowing on 38 weeks of corrected age.
In this case, the twins' symptoms were probably associated with both prematurity and congenital DM1. We speculated that the factor of premature birth made symptoms such as apnea and hypotonia of congenital DM1 worse, but we were not able to find the true prognostic factors from premature babies with congenital DM1. A previous study by Lee et al. (10) reported that a 30-week neonate with congenital DM1, who was initially ventilated for 55 days, survived and was free of respiratory complications by the age of 1 yr. In our case, the first and second twins were ventilated for 89 and 38 days, respectively. Second twin was able to breathe spontaneously relatively earlier, but apnea and chylothorax in the first twin delayed the weaning of ventilator therapy. It is well established that ventilation for more than 4 weeks is a significant prognostic factor in congenital DM1 of full term neonates (9). However, for the prognosis of congenital DM1 of premature babies, duration of ventilation should be more individualized and objectified through further studies of premature congenital DM1 patients.
In our case, the first twin characteristically showed chylothorax with non-immune hydrops. Pleural effusions with or without hydrops were frequently described in cases of congenital DM1 (11-13). According to literature, there were two cases in which chylothorax were associated with congenital actin myopathy and X-linked myotubular myopathy (14, 15). However, there is no information on the association with congenital DM1 and chylothorax. To our knowledge, this is the first report on congenital DM1 which accompanied the chylothorax. An X-linked recessive transmission of chylothorax has been suggested (16). More investigation on the association with chylothorax and congenital DM1 and other myopathies is needed.
De Andoin et al. (17) presented the case of congenital DM1, in which the mother conceived by IVF presented asymptomatic late onset form of DM1 that was ultimately diagnosed as a result of the condition inherited by the babies. It is reported that patients of both sexes with any form of DM1 tend to have impaired fertility (18). Obstetric risks like ectopic pregnancy, placenta previa, postpartum hemorrhage, preterm labor, polyhdroamnois, and perinatal mortality is markedly increased in pregnant patients with myotonic dystrophy (19). For this reason, it is not uncommon that undiagnosed women affected by DM1 are included in IVF programs and give birth to congenital DM1 babies. A previous study showed that preimplantation genetic diagnosis resulted in good pregnancy outcome for DM1 patients (20). In summary, it is important to carefully observe the clinical manifestations and genetic nature of women at childbearing age and diagnose DM1 before contemplating pregnancy.
In conclusion, we report the case with a rare IVF premature congenital DM1 twins. It is a rare case that congenital DM1 happened to twins and both twins survived at least 100 days after birth, even though they were premature babies. We would like to suggest that, with a case of severe neonatal hypotonia, congenital DM1 should be differentiated by DNA analysis in any gestational age. We recommend to consider that chylothorax is one of the clinical manifestations in DM1. Finally, since DM1 is a known cause of infertility and one of the most frequent adult myopathies, we should consider DM1 in infertility clinic with detailed history and physical examination.
 XML Download
XML Download